
ENTER
Seawater co2 Exstraction
The global gorilla intends to use this technology
Saline Water-Based Mineralization Pathway for Gigatonne-Scale CO2 Management
Erika Callagon La Plante, 112222Dante A. Simonetti, 11222Jingbo Wang, 11222Abdulaziz Al-Turki, 11222Xin Chen, 11222David Jassby, 112222and Gaurav N. Sant*
ACCESS
Metrics & More
Article Recommendations
ABSTRACT: This perspective proposes a potential pathway to diminish atmospheric CO2 accumulations which is distinct from traditional carbon capture and geological sequestration strategies and from existing negative emissions technologies (NETs). Unlike conventional sorbent- or solvent-based CO2 capture processes where substantial energy expenditures are associated with demixing and desorbing CO2, the single-step carbon sequestration and storage (sCS2) approach relies on electrolytic carbonate mineral precipitation using renewable energy within a simple and scalable process design.
Although numerous approaches have implied electrolysis for carbon management, the sCS2 approach is unique in the following ways:
(1) CO2 mineralization for promoting solid carbonate formation: The thermodynamic and kinetic barriers to carbonate precipitation are overcome by direct and in situ electrochemical forcing to stabilize dissolved inorganic carbon and divalent cations [Ca,Mg] to form carbonate minerals.
(2) Flow-through membraneless electrolysis: A flowing electrolyte (seawater) is dissociated while in motion. The process utilizes cost-effective mesh electrodes while also decreasing the number of components and assembly steps and reducing the risk of device failure.
(3) Integrated electrolytic reactor−rotary drum filter: An electroactive thin- film mesh cathode (eTFC) is suggested to be integrated within a rotary drum filter configuration, allowing for the filtration of dilute and polydispersed mineral precipitates at a low energy cost. These attributes render sCS2 as an approach worthy of more detailed evaluation, development, and scaling for global-scale carbon management.
KEYWORDS: Electrochemistry, Carbon dioxide, Climate change, Seawater, Mineralization, Renewable energy, Water treatment
INTRODUCTION AND BACKGROUND
More than 37 Gt of carbon dioxide (CO2; gigatonnes) is emitted globally per year from fossil fuel combustion and industrial processes.1 The reduction and, in time, the elimination of anthropogenic carbon dioxide emissions and the removal of accumulated carbon dioxide from the atmosphere are necessary to limit the increase in the global average temperature to 1.5 °C in 2100 to mitigate ongoing climate change.1,2 The emissions
gap for this target is on the order of 35 Gt in 2030.1 Furthermore, 10−20 Gt of CO2 will need to be removed from the atmosphere, annually (i.e., 72 to 144 × 106 mol/s), starting in 2050.3 This requires transformative solutions to “manage carbon” globally. The injection of CO2 captured from point sources or the
700 Mt per year over 50 years of injection.5 Although the conceivable capacity of geological sequestration sitesonshore and offshoreis anticipated to be more than sufficient to accommodate current (and future) levels of CO2 emissions, the risk of CO2 migration and leakage and the management and verification of the injection process necessitate significant monitoring of the wells, the subsurface, and the ground surface over time.6 In addition, traditional approaches for carbon management based on carbon capture, sequestration, and storage (CCSS) are hostage to (i) the thermodynamic penalties associated with the need to overcome the entropy of demixing CO2 from either air or a flue gas stream and the subsequent need to fulfill the enthalpy of desorbing CO2 from a solid or liquid
atmosphere into geological formations including (depleted) oil and gas reservoirs, unmineable coal beds, and saline aquifers could sequester up to 22,000 Gt of CO2 in North America.4 Although the theoretical capacity is enormous, practically, pressure limitations, needed to prevent rock fracturing or the reactivation of existing faults, and/or the presence of residual hydrocarbons result in a more modest storage capacity of around
© 2021 American Chemical Society 
substrate and (ii) the need for tremendous logistics and conveyance infrastructure (e.g., pipelines, compression stations, etc.) to transport CO2 to geological sequestration sites. Particularly, in conventional sorption−desorption-based CO2 capture, energy expenditure is associated with the separation of CO2 from a gaseous mixture, involving a decrease in the system’s entropy, and the desorption step which allows the concentration of CO2 to a grade sufficient for pipeline transport and, subsequently, geological sequestration. Taken together, while technical challenges remainand are indeed being progres- sively resolved−practical realization of CCSS is strongly (solely?) conditioned on supportive policy that empowers and derisks, and, pending best practices and time-bound monitoring, in the limit, holds harmless developers of CCSS projects around the world. Beyond geological sequestration and storage, changes in land use, agricultural practices, marine geoengineering, and
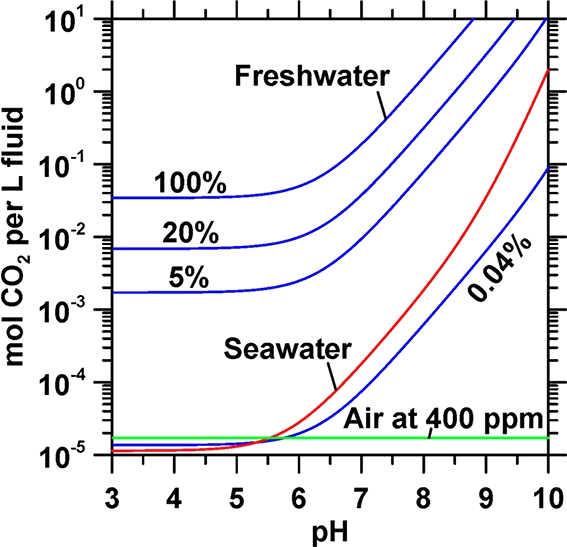
Figure 1. Total dissolved carbon content as a function of pH for solutions in equilibrium with gaseous streams of CO2 across a range of gas-phase concentrations. The total dissolved CO2 is equal to the transformation of CO2 chemicals.
to construction materials and
[H2CO3*] + [HCO −] + [CO32−]. For reference, 0.04% represents
the concentration of CO2 in air. The CO2 content in air is shown for
er alternate large-scale pathways that makeup our
comparison.
emerging portfolio of approaches for ensuring carbon manage-
ment (i.e., emissions reduction, and atmospheric carbon removal).3,7,8 Although some progress has been made in the development of negative (CO2/carbon) emissions technologies (NETs), much more substantive “exponential” advancements are needed to achieve the necessary rates of CO2 removal and durable carbon storage in a cost-effective/-viable manner.3 As one example, today, despite its enormous CO2 storage potential, our limited understanding of ocean chemistry and natural feedback cycles and the prevalence CO2/energy-intensive methods of production of consumable additives (e.g., acids and bases) preclude the widespread and rapid implementation of marine geoengineering solutions.
Over geological time scales, CO2 is fixed within stable mineral
[Ca,Mg] carbonates via biogenic and abiogenic processes. Inorganic carbon mineralization can occur through the formation of travertine deposits in surface waters and hot springs. For example, hyperalkaline Ca-rich groundwaters derived from the dissolution of ultramafic and mafic rocks in ophiolitic formations, upon reaching the surface, react with atmospheric CO2, precipitating calcium carbonates. Another mechanism of travertine formation involves geothermally heated alkaline waters with elevated pCO2, which upon reaching the surface degas to atmospheric equilibrium, leading to an increased pH and carbonate precipitation. In modern oceans, calcite and aragonite, which are polymorphs of calcium carbonate, are produced by coccolithophores (single-celled algae which form calcareous plates), foraminifera (single-celled protists), mollusks, echinoderms, and corals. Although carbo- nate formation in the ocean results in the release (“degassing”) of dissolved CO2 because of the consumption of alkalinity9 (i.e., because the CO2 storage capacity enhances with alkalinity; see Figure 1), carbonate mineral precipitation if induced via externally forced alkalization could result in net CO2 abatement. This premise forms the basis of our saline water (“seawater”)- based mineralization approach.
New Concept. We propose an engineered/synthetic carbon
management solution that has the potential for implementation at a global scale and which may be capable of much higher rates of carbon mineralization and CO2 removal than existing NETs. The basis of our single-step carbon sequestration and storage (sCS2) strategy involves forcing the rapid precipitation, electrolytically, of calcium carbonate (CaCO3), magnesium carbonate (MgCO3), hydroxy-carbonates, and their variants by combining dissolved CO2 (i.e., solubilized in seawater and other
water bodies to a level described by Henry’s Law which establishes the ocean−atmosphere equilibrium vis-a-̀vis CO2 and other gases) with Ca2+ and Mg2+ species in an aqueous (reaction) medium using electrolytic flow reactors. For context, modern oceans are oversaturated, on average, with respect to CaCO3, and MgCO3 by a factor of more than 2 times.10 The rationale for removing CO2 from the atmosphere (“air”) via
water is straightforward: water contains nearly 150 times more CO2 than air per unit volume (Figure 1). Thus, the approach exploits mineralization reactions to achieve carbon sequestra- tion and storage, while bypassing the capture (“CO2 concentration”) step.
Importantly, this approach does not require overcoming the substantial entropy of demixing CO2 and simultaneously exploits the favorable thermodynamics of carbonate precip- itation from alkaline solutions. The sCS2 concept, thus, specifically (a) minimizes process complexity and emphasizes modularity (i.e., by minimizing the number of discrete unit operations) and (b) emphasizes process intensification (PI) by renewable energy integration. With its ability to encompass a wide-range of CO2 concentrations, the sCS2 approach enables flexibility in achieving carbon management across diverse locations and settings without the need for complex logistics facilities (e.g., pipelines, compression stations, etc.) that would be required for traditional CCSS solutions.
Although the controlled sequestration of CO2 within carbonate minerals is a well-known process that has been extensively studied and even demonstrated at an industrial scale, several attributes of the sCS2 process distinguish it from existing carbonate-formation strategies. Foremost, as opposed to deep injection-based in situ mineral trapping processes (e.g., CarbFix11) which require year(s), the sCS2 approach achieves ex situ mineral storage of CO2 via carbonate mineral formationat the Earth’s surfacewithout the need for concentration, compression, and injection of CO2, within seconds to minutes. Second, unlike ex situ mineral carbonation approaches which convert CO2 to carbonates,12,13 the sCS2 process is not mass limited by the availability and rate limited by the reactivity of the feedstocks (e.g., alkaline wastes). For
example, sCS2 does not rely on the slow dissolution of alkaline rocks (≈10−12 to 10−8 mol/m2/s depending on the temperature and chemistry of the solvent14−18), which limits the rate of CO2 mineralization that can be achieved unless vigorous pretreat-
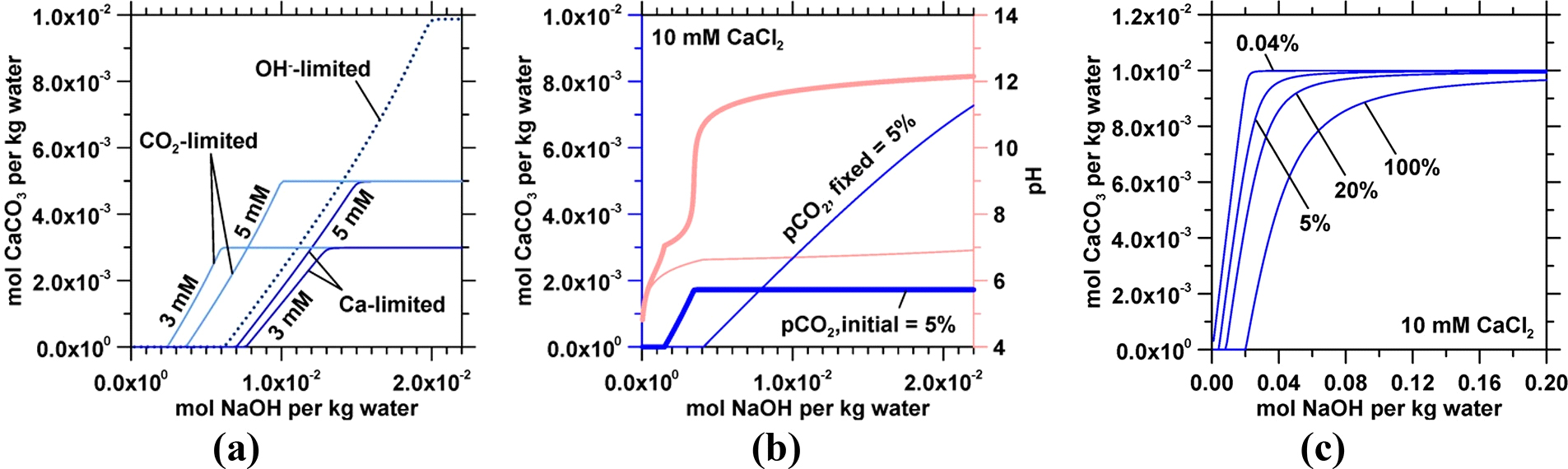
Figure 2. (a) Representative calculations demonstrating the limits on calcite precipitation as affected by Ca, CO2, or alkalinity for a solution of composition [Ca] = 10 mM, [Cl] = 20 mM, [CO2] = 10 mM (∼30% CO2; 300,000 ppm), and pH 4.16 (dotted curve). The maximum CaCO3 yield is 10 mmol per kg water. If [Ca] is decreased to 3 or 5 mM (dark blue curves) ([Cl] is either 6 or 10 mM), [CO2] = 10 mM, and pH 4.17, the maximum CaCO3 yield decreases to 3 and 5 mM. Similarly, decreasing [CO2] to 3 mM (light blue curves) (∼9% CO2, pH 4.42) or 5 mM (∼15% CO2, pH 4.31) ([Ca] = 10 mM, [Cl] = 20 mM) decreases the maximum CaCO3 yield. Interestingly, where decreasing [Ca] leads to an increase in NaOH consumption for an equivalent CaCO3 yield, decreasing [CO2] decreases NaOH consumption. (b) Two scenarios are compared: (i) the initial pCO2 is 5%, corresponding to 1.73 mM CO2 (bold curves), and allowed to decrease with precipitation and (ii) the solution’s pCO2 is held constant (e.g., by continuous equilibration with a gaseous CO2 stream at 17,300 ppm) at 5% (thin curves). In (i), CaCO3 precipitation is induced rapidly by NaOH addition and is limited by the total dissolved CO2 as in (a). The continuous supply of CO2 in (ii) enables CaCO3 precipitation at a yield of around 7 mM (limited by [Ca]). (c) NaOH consumption for calcite precipitation for a solution in equilibrium with CO2 at different pCO2 levels (in vol %) as dictated by Henry’s Law (herein, in all cases NaOH is simply used to indicate the effect of solution alkalinity).
ment (grinding) to enhance feedstock reactivity and elevated pressure and temperature are applied to realize appreciable kinetics. These issues are obviated in the sCS2 approach since it utilizes Ca and Mg that are already solubilized in seawater. In addition, mineral carbonationespecially when marginally reactive feedstocks are usedoften requires concentrated CO2 streams, whereas, as is elaborated below, the sCS2 approach can be applied without any cost escalation over a wide range of CO2 concentrations (i.e., atmospheric concentrations to 100 vol % CO2) and temperatures (i.e., ambient to ∼90 °C). Third, and
finally, the sCS2 process on account of inducing in situ alkalinization, electrochemically, does not require solvents, buffering agents, or stoichiometric additives to induce CO2 absorption or solvent alkalinization.19−21 This is important as the production of stoichiometric additives (e.g., bases such as
NaOH, NH4OH, etc.) is both CO2 and energy intensive, a major deficiency of any potential NET which requires such additives for CO2 mitigation.
Numerous approaches for carbon management are based on electrochemical stimulation, for example, involving the elec-
trolysis of water. The basic premise involves the generation of
architecture of electrolyzers, the sCS2 process mimics flow reactors to maximize yield and facilitate upscaling and parallelization, which is more difficult to accomplish with electrodialysis systems.24,25 Lastly, while existing approaches have often sought to induce bulk pH shifts of the entire fluid volume,28−30 the sCS2 process generates elevated alkalinity in small control volumes, for example, in the vicinity of the mesh
electrodes where the driving force for precipitation is maximized, to promote energy efficiency and to ensure precise reaction control.
METHODS
Thermodynamics and Kinetics of Carbonate Precip- itation. The precipitation of calcium carbonate, for example, calcite, is given by31
CaCO3 (calcite) F Ca2+ + CO32−, log Ksp = −8.48 at 25° C
(1)
where Ksp is the solubility product (also known as the equilibrium constant) and is equal to the product of the
aqueous activities of Ca2+ and CO 2− at equilibrium. During the
acidity and/or alkalinity while emphasizing processes that can be
3 precipitation of calcite, either HCO3−
or CO 2−
,both formed
operated using renewable energy. This prominently includes approaches based on the classical chloralkali process,22,23 for example, for the production of chemicals, or electrodialysis, for example, to enable indirect ocean capture (IOC), of CO2 as either a gas or a mineral. However, issues including (a) the tremendous energy intensity of chloralkali style processes, (b) the need for flue gas borne CO2, and/or (c) fouling and
through the speciation of CO2 in water, may adsorb and incorporate on the growing surface.32 The Ksp of CaCO3 decreases with temperature, such that raising the temperature of a calcite-saturated solution from 25 to 90 °C results in the precipitation of calcite with a yield of around 300 μmol/kg of
water. The speciation reactions and dissociation constants that describe the CO2−H2O system are written as
degradation of ion-exchange (or bipolar) membranes24,25 that are required for electrodialysis; for example, when the feedwater
H2CO3* F HCO − + H+, log K1
= −6.35
(2)
contains divalent cations due to the formation of slightly soluble carbonates, remain problematic.22,26,27 Although the sCS2
HCO − F CO 2− + H+, log K2
= −10.33
(3)
process also exploits electrochemical forcing (of carbonate mineral formation), it is uniquely differentiated, as it does not require typical fine-pore membranes that are prone to fouling and scaling. Rather, it utilizes flow-through coarse-mesh electrodes. Moreover, unlike the more common static
where H2CO3* denotes the total CO2 (aq) and H2CO30. The partitioning of dissolved CO2 is disclosed by a Bjerrum diagram. In general, the activity of CO32− anions depends on pH (e.g., in water, CO 2− is the dominant carbon species at pH > 10.33) and so does the extent of calcite precipitation. Higher salinities shift
K1 and K2 to greater values, and thus, the pH where HCO − and CO32− ions are dominant shifts to lower values. The thermodynamic driving force for mineral precipitation is given by the saturation ratio, Ω = IAP/Ksp, where IAP is the ion
activity product; for example, for calcite, this is the product of the activities of Ca2+ and CO 2− in solution. This is of relevance to natural waters (e.g., groundwater, seawater, produced water) that contain divalent metal cations since their circumneutral pH requires the provision of supplemental alkalinity to induce
carbonate precipitation. Because Ca and Mg are the most abundant divalent cations in natural waters and often industrial waters (e.g., produced water, thermoevaporation brines, etc.), in the examples that follow, we represent such waters as CaCl2
solutions since dominantly Cl− offers charge compensation to
the cations in these systems. For the sake of simplicity, we focus our analysis on calcite, the most thermodynamically favorable crystalline calcium carbonate polymorph and one that is abundantly represented in nature. The general analysis frame- work herein applies analogously to magnesite, hydrous carbonates, hydroxy-carbonates, and mixed-metal carbonates (e.g., magnesium-rich calcite), although it is recognized that the formation of magnesium carbonates may be subject to more serious kinetic controls on precipitation.33,34 We consider the nonideality of solutions; thus, the geochemical calculations consider the ion activities explicitly.
A straight forward (although unfeasible) approach for over- coming the kinetic barrier to carbonate precipitation in seawater involves the addition of NaOH as a stoichiometric additive to raise the solution’s pH and Ωcalcite, resulting eventually in calcite’s precipitation. For a solution of a fixed initial CO2 concentration (pCO2), calcite precipitation is limited either by the abundance of Ca, CO2, or alkalinity (pH). These scenarios are illustrated in Figure 2(a), which considers a CaCl2 solution with added CO2 at various concentrations of Ca, CO2, or alkalinity (i.e., as modeled by the addition of NaOH). Although less acidic solutions (i.e., having lower CO2 concentrations) require smaller amounts of NaOH to initiate calcite precip- itation, the maximum CaCO3 yield, which in this scenario is limited by CO2, will accordingly be lower (light blue curves in Figure 2a). On the other hand, excess CO2 in solution will render Ca the limiting reactant. The case where both NaOH:CaCO3 and H2O:CaCO3 molar ratios are minimized is one in which Ca and CO2 molar concentrations are approximately equal, and NaOH is added until the maximum CaCO3 yield is achieved (dotted curve in Figure 2a). The addition of NaOH results in the complexation of Na+ with
CO 2−; thus, the molar ratio between CO2 and Ca to reach
scenario, precipitation is initially inhibited because of excess acidity, that is, CO2. For completeness, different scenarios of CO2 equilibrium and replenishment for varying pCO2 levels are shown in Figure 2(c). In the limit, the carbonate yield is dependent only on the feed’s [Ca] abundance and is invariant with the CO2 concentration (see also Figure 3).
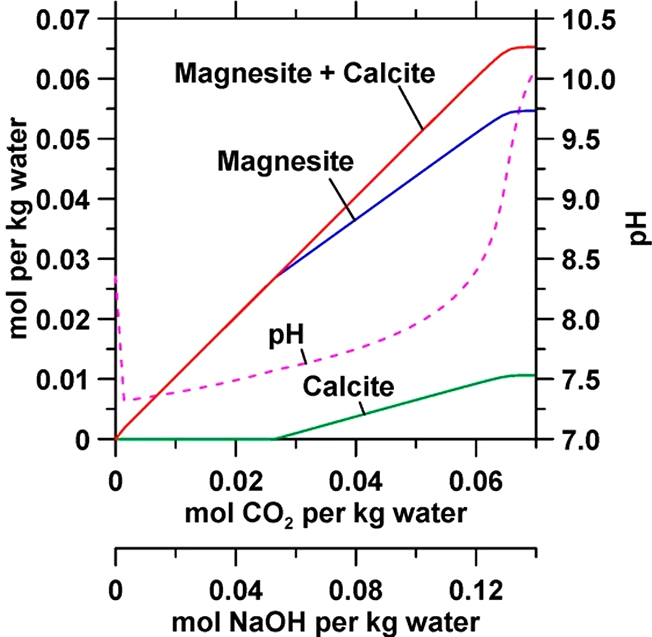
The amount of Figure 3. Representative equilibrium calculations using PHREEQC75 with the llnl.dat database for a reference seawater composition as given by Millero et al.10 The simultaneous addition of CO2 and NaOH at a 1:2 molar ratio results in the precipitation of calcite and magnesite, up
to about ∼55 mmol magnesite and ∼10 mmol calcite per kg water. The CO2 saturation concentration at atmospheric pressure is ∼34 mM. In
an engineered process, CO2 equilibrium can be maintained by simply bubbling air over the course of carbonate precipitation (Figure 2b, c).
CO2 that can dissolve in water is controlled by its pH and the salinity- and temperature-dependent Henry’s law constant. For a given solution, increasing the solution’s pH (pH > 6) increases total dissolved carbon (Figure 1). This is on account of the pH- dependent speciation of CO2(aq) to HCO − and CO 2−, which reduces the concentration of CO2(aq), allowing for further dissolution of CO2 (g) as per Henry’s law. It is for this reason that the formation of cation-carbonate and cation-bicarbonate
complexes in seawater increases its carbon storage capacity relative to freshwater at pH > 6.35,36
Carbonate precipitation reactions are characterized by a time scale.37 Under well-mixed conditions (i.e., free of mass transport limitations) at 25 °C and 1 atm, the equilibrium described by reaction 4 occurs within t = 5.0 × 10−11 s.37
CO2(g) F CO2(aq)
(4)
The aqueous species H CO , HCO −, and CO 2−, as described
3 2− 2+
2 3 3
3 37
equivalence between {CO3 } and {Ca }, where brackets {}
by reactions 5−7, reach equilibrium within 10−2 s.
denote the activity, is slightly higher than 1. The molar ratio between NaOH consumed and CO2 stabilized as CaCO3 is at
CO2(aq) + H2O F H2CO3
(5)
the minimum 2 (i.e., which offers one mole of OH− for each of
reactions 2 and 3).
H2CO3 F H+ + HCO −
(6)
For waters which feature low CO2 concentrations, air can be bubbled through them to maintain CO2 abundance in relation to the rate of CO2 consumption. To illustrate this, two scenarios are compared in Figure 2(b): one in which the initial CO2 concentration is fixed and dissolved carbon is progressively
HCO − F H+ + CO 2−
(7)
However, equilibrium with respect to Ca2+ (i.e., reactions 8 and 9) is only reached in 103 s.37
Ca2+ + CO 2− F CaCO (aq)
depleted and another in which the solution’s CO2 concentration 3 3
(8)
is held constant by equilibration with a gas stream which features a fixed pCO2. In the first case, CaCO3 precipitation is induced
CaCO3(aq) F CaCO3(s)
(9)
rapidly by NaOH addition and is limited by the total dissolved CO2, whereas in the second case, the replenishment of CO2 enables calcite precipitation until Ca depletion. In the second
In alkaline solutions (pH > 10), the alternative pathway of CO2 solvation by reaction with OH− to form HCO − is even faster (k
= 8.5 × 103 M−1 s−1) than that with H2O (k = 6.6 × 10−4 M−1
s−1).38,39 In support of these estimates, our own data of calcite precipitation rates in concentrated solutions similar to seawater (i.e., simulated “produced water” containing greater than 1 M NaCl and impurities of magnesium, sulfate, and others) indicate a precipitation rate constant on the order of 3.2 × 106 M s−1, with a yield that is consistent with Figure 2(c).40 This rate constant is derived by fitting experimental (calcite precipitation) data using an equation of the form R = k(Ω − 1)n,41 where R is the precipitation rate in M s−1, k is the rate constant in M s−1, Ω is the saturation ratio with respect to calcite, and n is the reaction order.
Energy Intensity Analysis. The energy demand of a mineralization process that uses seawater as a source of divalent cations and NaOH as a stoichiometric additive can be estimated for comparison with a geological CCSS strategy (Figure 4) and
whereas the current typical value used in the baseline case is 1.3 kWh per t of CO2 mineralized per m of total dynamic head42), it is instructive to use data from operating desalination plants that rely on seawater as their source water, much like both the baseline and proposed sCS2 process. A robust review of existing plants reveals that the energy required to provide seawater to a 50 MGD (million gallons per day) plant is 0.45 kWh/m3.43 On the basis of this estimate and the assumption that the vast majority of sCS2 plants would be built on the coast, we estimate
the energy required by seawater pumping to be 159 kWh/t CO2 mineralized. Chemical dispersion requires between 2.8−7.7 kWh per t of CO2 mineralized, and sedimentation requires 0.18 to 0.35 kWh per t of CO2 mineralized.44 Thus, in total, water processing and handling will consume on the order of 100 kWh
per t of CO2 mineralized assuming a seawater feed. Furthermore, the energy for water processing can be further decreased by operating directly in the open ocean (e.g., using ship-based or freestanding platforms).45,46 The synthesis of NaOH by the chlor-alkali process requires 2.5 MWh per t NaOH.47 Therefore, we estimate that the energy demand for direct CO2 mineralization using NaOH as an additiveand using seawater as the source of both divalents and (solubilized) CO2to be on the order of 4.5 MWh per t CO2, exclusive of water processing. Thus, the associated cost of CO2 removal estimated from the price of electricity for industrial use of about $70 per MWh48 is
$315 per t CO2 for current best-in-class chloralkali-produced
NaOH. The need for NaOH may be somewhat reduced by carbonating alkaline solids, whose dissolution produces alkaline
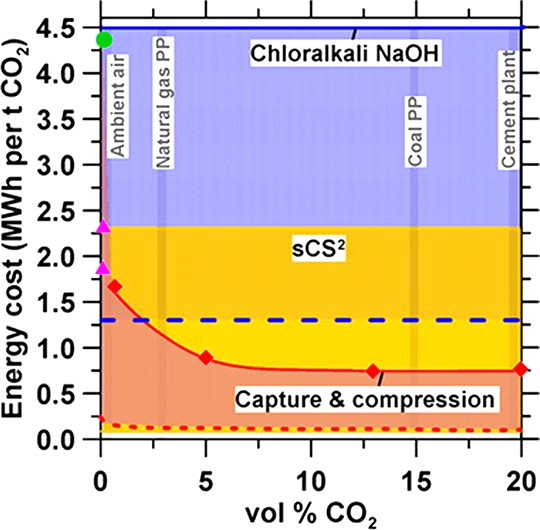
Figure 4. Energy requirements for CO2 capture and compression as a function of concentration for an amine-based process simulated using Aspen Plus (solid red curve). The thermodynamic minimum energy (dotted red curve) required for CO2 separation from a mixture and compression from 1 atm to 15 MPa is calculated based on the entropy of (de)mixing gas phase CO2. Also shown are the energy costs for CO2 mineralization (solid blue line) for chloralkali-produced NaOH (dashed blue line) at the thermodynamic minimum energy demand for production. The theoretical energy requirement for NaOH production from NaCl is taken from Thiel et al.51 The energy costs for direct air capture and compression (DACC; magenta triangles) using a KOH/K2CO3 process and integrated caustic-amine (green circle) are taken from Keith and co-workers, refs 93 and 94, respectively. The vertical gray lines represent CO2 concentrations in air, natural gas- and coal-fired power plants, and cement plant flue gas. The shaded areas represent representative ranges of energy cost for the following CO2 mitigation strategies: (red) capture and compression (range: 0.1 MWh at the thermodynamic minimum to 4.5 MWh per tonne of CO2, depending on concentration), (blue) stoichiometric addition of NaOH (range: 1.26 MWh at the thermodynamic minimum to 4.5 MWh per tonne of CO2), and (yellow) the proposed electrolytic precipitation approach (sCS2) exclusive of water intake (range: 0.07 MWh at the thermodynamic minimum to 2.3 MWh per tonne of CO2, independent of concentration).
other possible approaches. Unlike geological CCSS, in general, seawater mineralization-based CO2 abatement does not require a traditional CO2 capture step. Thus, the energy requirements of the baseline process (i.e., although practically infeasible on account of the consumable NaOH demand) are based around the needs of water handling and processing and NaOH production. Although the cost of pumping seawater is highly dependent on the location of the plant vis-a-̀vis the ocean, the type of intake, and the size and type of pipes used to move the water (e.g., process modeling indicates an energy requirement for water intake of 41 kWh/t CO2 for a 30 m dynamic head,
metals (Ca2+, Mg2+) and OH− in solution. The energy input for
such direct carbonation includes pretreatment costs, including grinding and, in some cases, thermal activation, (carbonate) product disposal, and the operation of pumps and mixers, is on the order of 0.5 MWh per t CO2, all inclusive.49 Unfortunately, such direct carbonation of industrial alkaline solids is expected to deliver no more than 1.2 Gt of CO2 abatement per year.50
A somewhat less energy intensive pathway of NaOH production may be achieved by bipolar membrane electro- dialysis.51 Even if it were possible to produce NaOH at its theoretical minimum energy demand of 0.7 MWh per t NaOH (45% NaOH with a coproduct, HCl, that can be used for enhanced silicate dissolution51), the cost of mineralization- based CO2 management would, at the minimum, be associated with an energy intensity of 1.26 MWh per t CO2, corresponding to a cost of no less than $90 per t of CO2 converted into solid carbonates using seawater-derived divalent cations. For comparison, based on the heat required for kiln operation (200 kJ/mol CaO) and the amount of CO2 sequestered per mol of lime (1.79 mol CO2/mol CaO),52,53 calcination-based pathways of inducing alkalinity require at least 0.7 MWh per t CO2 sequestered in dissolved form, although this does not include the additional energy demands of CO2 capture, compression, and storage, an obvious requirement.
We estimated the energy intensity of the traditional CCSS
pathway by considering a monoethanolamine (MEA)-based process consisting of an absorber, stripper, cooler, and four-stage compressor using Aspen Plus54 with the eRNTL thermody- namic property method (Figure 4).55,56 Herein, CO2-depleted gas was assumed to be extracted from the top of the absorber, while the CO2-rich solvent stream is extracted from the
bottom.57−59 To release CO2, the CO2-rich amine is heated to
fulfill the enthalpy of desorption. An overhead condenser provides a reflux liquid stream to the column and purifies the CO2-rich gas to nearly 100% CO2. The near pure CO2 stream
released from the stripper is compressed and transmitted for geological storage. Considering an inlet stream with 3% CO2 (e.g., corresponding to the flue gas emitted from a natural gas fired power plant), an energy intensity of ∼1.5 MWh per t of CO2 is estimated for amine-based CO2 capture (heat duty for amine regeneration of 1.3 MWh per tonne) and for
pressurization of the recovered CO2 stream (0.2 MWh per tonne) from atmospheric pressure to a pipeline specification at 14 MPa. This energy intensity decreases to 0.8 MWh per tonne CO2 as the concentration of CO2 in the inlet stream increases to
∼12% (for a coal-fired power plants; 0.6 MWh for carbon
capture, and 0.2 MWh for compression per tonne of CO2), remaining constant thereafter. However, at inlet CO2 concen- trations below 3% CO2, the energy intensity of an amine-based process sharply increases primarily because of the increase in the heat energy required to desorb CO2 from the MEA solvent at
typical chloralkali-based NaOH production and amine solvent- based processes at ambient concentrations of CO2 (i.e., conservatively, less than 2.3 MWh per tonne versus greater than 4 MWh per tonne). (ii) If the energy benefit of the hydrogen coproduct is considered and/or zero-carbon energy input is used, in each case, the sCS2 approach offers a basis for a true NET, without a need to redirect renewable electricity to displace traditional fossil fuel-based power generation (further discussion below).
Cost Estimates. At a first approximation, the capital expenditure (CapEx) and the levelized cost of CO2 abatement (LCCA, $/t) can be estimated starting from today’s costs of alkaline electrolyzers for a full-scale commercial plant which abates 1 million tonnes of CO2 per year (i.e., mineralizing 1 t CO2 will produce, at minimum, 2 t of carbonates and require 350
t of seawater). For example, today it is estimated that alkaline
low loadings.60 The low capacities of amines (∼0.25 mol per
mol),61 for atmospheric CO2 capture, would require more than
electrolyzers cost around $430 per kW.67 Since sCS2 does not
5.0 MWh per tonne CO2 of reboiler duty to achieve less than
0.05 mol per mol of working capacity.60 This indicates a particular advantage of mineralization processes to be less energy intensive, operationally speaking, for cases where CO2 feed streams are exceptionally dilute, such as in the removal of CO2 from the atmosphere or atmosphere-equilibrated water.
Also shown in Figure 4 is the energy requirement for the proposed electrolytic sCS2 mineralization process based on in situ electrochemical OH− generation that can be estimated based on current state-of-the-art near-commercial electrolyzers operating at 79% efficiency (i.e., 50 kWh of electricity to
generate 1 kg of H2 assuming a thermodynamic demand of 39.4 kWh/kg for the stoichiometric hydrogen evolution reaction: HER).62 Herein, 1 kg of H2 produced via the electrolysis of water yields 1000 mol of OH− ions which can sequester, on a stoichiometric basis, 22 kg of CO2 for an energy intensity of 2.3
MWh per tonne CO2. If we consider the calorific value of the coproduced low-pressure hydrogen [H2(g)], assuming a conversion efficiency (i.e., to combust H2(g) and produce electricity) on the order of 60%,63,64 similar to natural gas combution, the process yields an energy intensity of 1.2 MWh per tonne of CO2 mineralized. This analysis considers a reaction
stoichiometry where 2 mol of OH− mineralize 1 mol of CO2 into
calcium carbonate (CaCO ). Following this basis, 45 kg of low-
imply a traditional electrolyzer, we amplify our own CapEx estimate by 1.5 for a component CapEx of $650 per kW. For assumptions on the blended uptime of the plant (90%), the balance of plant costs of 50% of total CapEx (inside battery limit: ISBL, e.g., including pumps, piping, etc.), remainder of costs, which is owner’s cost + outside battery limit (OSBL) (20% of
ISBL) cost, low-pressure hydrogen yield (45 kg per t CO2 valued at $2−3 per kg H2), average financing costs (10% of total CapEx (ISBL + OSBL)), capital recovery factor (12.5%), and cost of electricity ($30 per MWh, i.e., consisting of renewable solar, wind, with or without storage),68 the analysis yields an “all-in”
gross capital cost on the order of $500 per tonne of CO2 mineralized and operational expenditure (OpEx, i.e., including energy costs and fixed operations and maintenance costs) on the order of $83 per tonne of CO2 mineralized. This yields LCCA(gross) = $145 per tonne of CO2 mineralized (i.e., including CapEx and OpEx, while applying a capital recovery factor to the CapEx component) when no cost offset is drawn from the sale of H2(g). However, if the “green hydrogen” produced were to be sold in commercial markets, in its low-cost low-pressure form, the cost set that results yields LCCA(net) = $55 per tonne of CO2 mineralized (H2 = $2 per kg) or $10 per tonne of CO2 mineralized (H2 = $3 per kg). Conservatively, no value is
pressure H 3
2(g) would be generated for every tonne of CO2
allocated to the carbonate mineral produced or production of
softened water that reduces the energy need of downstream
mineralized. Such green hydrogen is expected to offer a commercial value on the order of $3/kg,65 such that a cost offset on the order of $135 could be realized per tonne of CO2 mineralized. On the other hand, if the low-pressure hydrogen produced were to be converted into electricity using a hydrogen fuel cell (HFC), a conversion efficiency on the order of 80% could be realized,66 and an overall energy intensity of 0.84 MWh per tonne of CO2 mineralized would result. The base energy intensity would further decrease to 1.9 MWh per tonne of CO2 (without H2 recovery; $133 per tonne of CO2) and 0.38 MWh per tonne CO2 (with H2 recovery and conversion at 90%
efficiency using a HFC; $27 per tonne of CO2) for an
desalination operations (if any).
Potential Effects on Seawater Chemistry. The implica- tions of changes in the prevailing abundance of Ca and Mg in seawater can be significant. For example, if changes in divalent cation concentrations result in a change in solution pH, this alters the carbon storage capacity of seawater (Note: In effect, increasing alkalinity enhances CO2 storage in seawater and vice versa; Figure 1). Using a reference seawater composition10 and a volume of 1.3 × 1021 L, the global ocean inventories of Ca and Mg are estimated as 1.4 × 1019 and 7.1 × 1019 mol, respectively.69 The removal of 3.6 × 1013 mol of Ca and 1.9 ×14
electrolyzer operating at 90% efficiency [Note: The thermody- namic minimum of the sCS2 process is on the order of 0.07 MWh per tonne of CO2 mineralized for atmospheric CO2 for an electrolyzer efficiency and H2-to-electricity conversion efficiency of 100%]. These values, collectively, bound the practical energy intensities for sCS2. Taken together, this analysis and Figure 4 indicate the following: (i) Direct electrolytic mineralization may be able to achieve carbon removal nearly twice as effectively as
10 mol of Mg per year will result in ∼0.03 mol % decreases in
total dissolved Ca and Mg. In a closed system, these reductions would result in a pH reduction of 0.05 units and in both log Ωcalcite and log Ωaragonite of 0.05 units after a century. As such, much more work is needed to ascertain the effects of approaches that seek to exploit the oceans for CO2 management and the implications of such approaches on affecting not only the ocean’s chemistry but also its fragile ecosystems.
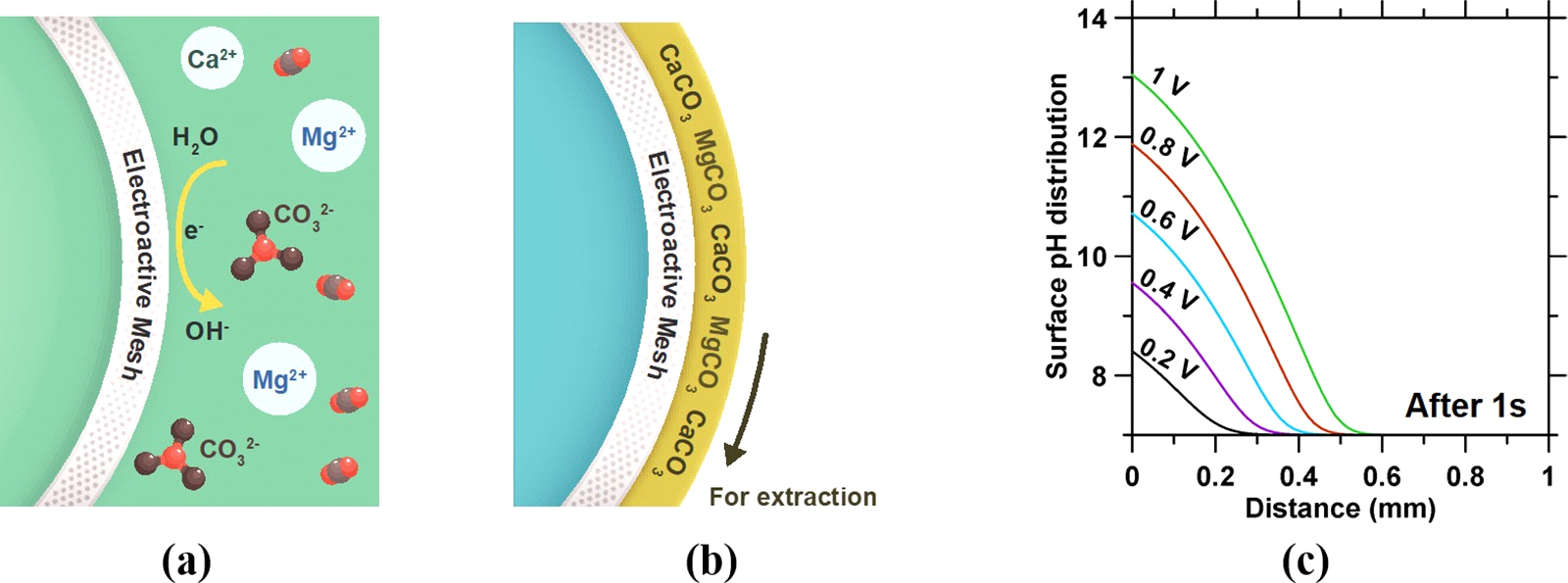
Figure 5. Conceptual illustration of (a) localized OH− generation on the (b) mesh cathode as a means to induce carbonate precipitation. The mass deposited on/near the mesh surface is removed using a rotory drum filtration approach. (c) pH of the electrolyte for different overpotentials 1 s after electrical polarization as simulated using COMSOL Multiphysics using adaptive time-stepping, triangular mesh elements (mesh opening of 173.21 μm2), and periodic boundary conditions. The breakdown potential of water is assumed as 0 VRHE (RHE: reversible hydrogen electrode).104 This simulation considers a planar electrode (100 mm2) that is composed of 304 L stainless steel mesh immersed in excess electrolyte (0.1 M NaCl) where for the hydrogen evolution reaction (HER) the Tafel relationship [η = 0.172 + log(i/i0), with η the overpotential (V), i is the current density (A/m2), and i0 is the exchange current density (1.04 × 10−7 A/m2)] indicates pH(t) = 14 + log[{(10[(η‑1.2)/0.172])t}/9.6485 + 10−7], where pH is the average pH generated in the proximate saline electrolyte within a region that is 1 mm thick, η is the overpotential (V), and t is time (s). For example, an overpotential of around 0.5 V is needed to generate a pH > 10 at the mesh surface at which all the inorganic carbon in solution is speciated in the form of CO32− anions. Expectedly, increasing the surface area (e.g., using a finer mesh) of the electrode or its electrochemical activity would reduce the
overpotential needed to induce near-surface alkalinization.
RESULTS AND DISCUSSION
Establishing a Baseline. In a well-mixed system with low
mass transport resistances, carbonate-mineral precipitation is rate limiting (see Methods).37 In general, our analysis indicates that in seawater, alkalinization helps overcome the thermody- namic barrier to carbonate precipitation. Thus, foremost, we examine a typicalalthough unfeasibleapproach of addition of a strong base such as NaOH to a circumneutral Ca- and Mg- containing solution (Figure 3). Seawater beyond Ca species and NaCl (the dominant salt), additionally, also contains dissolved organic matter, phosphate ions, magnesium ions, and sulfate ions, which impose kinetic restraints on carbonate precipitation that is often attributed to the blocking of reactive nucleation and growth sites.70−73 For this reason, although seawater is supersaturated by a factor of at least 2 times with respect to calcite, extensive abiotic calcite precipitation is not observed. Nonetheless, in such kinetically inhibited systems, once initiated, precipitation proceeds to equilibrium, with yields that are congruent with expectations from equilibrium calculations.74 These aspects are further discussed to create a strategy for overcoming the kinetic barrier to carbonate precipitation.
For reference, it should be noted that the CO2 concentrations from point source emissions are around 3−4 vol % for natural gas-fired power plants, 12−15 vol % for coal-fired power plants and iron and steel mills, 20−25 vol % for cement plants, and greater than 90 vol % from ammonia, ethanol, and hydrogen plants.76 On the other hand, the atmospheric CO2 concentration is around 0.04 vol %,77 whereas CO2 captured by amine scrubbing can be more than 99 vol % pure.78 Therefore, we offer reference concentrations of 0.04%, 5%, 20%, and 100% in our analysis. CO2 mineralization as envisaged in the baseline case is modeled by analogy to water treatment processes. Therefore, first, if CO2 sources other than air are to be used, effective mixing and CO2 equilibration with saline water can simply be achieved using aeration tanks similar to those used in activated sludge processing. Thereafter, NaOH (i.e., a consumable additive; baseline scenario) could be mixed into the CO2-rich water, as in
coagulation and flocculation processes, resulting in the precipitation of mineral carbonates. The precipitates are then separated from the solution by sedimentation, and the discharge solids can be dewatered using belt presses or discharged into the ocean similar to brine disposal in typical desalination operations. Ascertaining Calcium and Magnesium Sufficiency for Carbonate Mineralization. Foremost, Ca and Mg are available in more than sufficient quantities to meet the demands of global-scale carbon management. For example, the removal of 10 Gt of CO2 per year requires 9.1 Gt Ca or 5.5 Gt Mg, equivalent to 0.0017% of the total Ca and 0.00032% of the total Mg contained in the world’s oceans (“seawater”). Alternately, although at much smaller levels, calcium and magnesium can be sourced from the following: (a) Saline groundwater can contain more than 1000 mg per L of total dissolved solids (TDS), whose withdrawal rates in 2015 reached 3.2 billion m3 per year,79 corresponding to 0.6 Mt Ca and 0.3 Mt Mg (using typical Ca and Mg concentrations in brackish waters in the U.S.).80 (b) Desalination brines produced globally at a rate of 50 billion m3 per year81 can supply an additional 0.04 Gt Ca and 0.1 Gt Mg annually. (c) The generation of 2.23 Gt per year of produced water in the U.S. alone,82 assuming an average Ca concentration of 5000 mg per L,83 can provide an additional 0.01 Gt Ca per year. Although alkaline byproducts resulting from the manufacturing of metals, alloys, and cement, and from coal combustion are rich in Ca and Mg, their weathering too is postulated to fix no more than 1.2 Gt CO2 per year.50 All that said, seawater remains the most viable and abundant source of divalent cations for mineralization processes, so long as the
ocean chemistry is unperturbed.
From stoichiometry, the conversion of 1 mol CO2 to 1 mol CaCO3 (or MgCO3) requires 2 mol NaOH (Figures 2a and 3). Therefore, the mineralization of 10 Gt of CO2 requires, at the minimum, approximately 18 Gt NaOH. However, global production of NaOH is comparatively trivial, on the order of 70 Mt in 2016,84 although simply based on its Na content 3.2 × 107 Gt NaOH could be synthesized using ocean water. To meet this demand, we would need 6000 large chloralkali plants, each
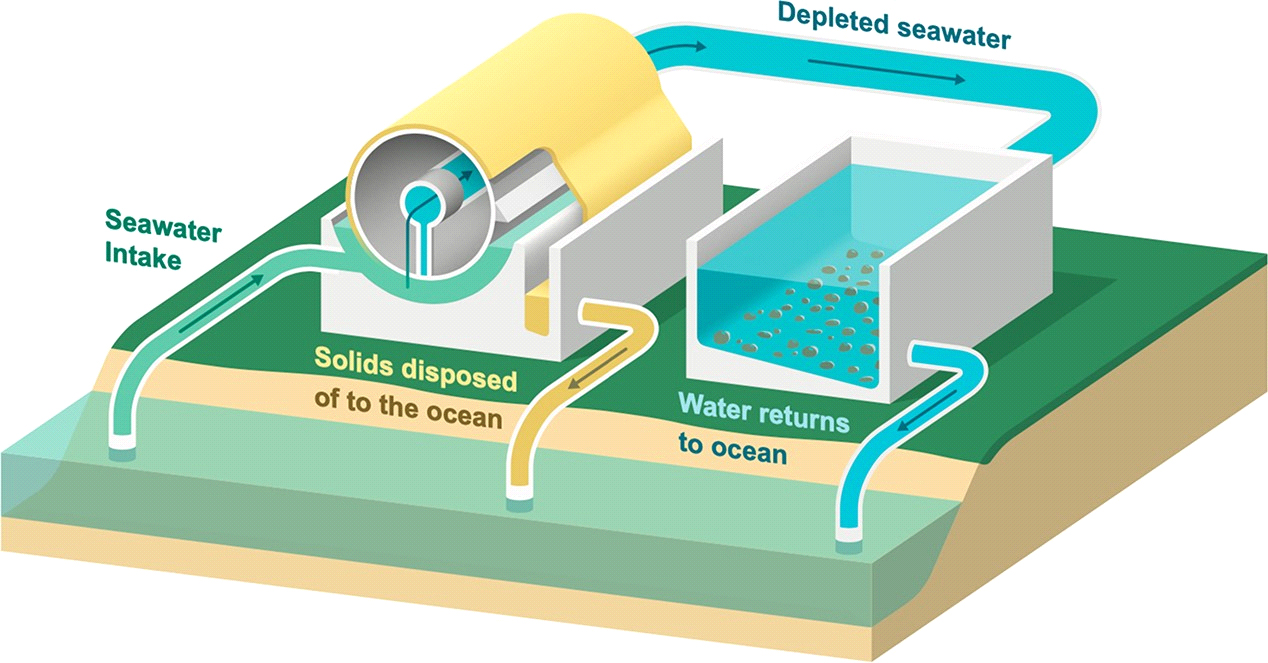
Figure 6. Conceptual illustration of the sCS2 process for achieving CO2 mineralization and disposal that utilizes an electrolytic flow reactor and integrated rotary drum filter concept to achieve rapid carbonate mineralization.
producing 3 Mt NaOH annually, an unfeasible proposition.85 The dosage of NaOH in sufficient quantities to seawater ([Mg]
≅ 55 mM, [Ca] ≅ 10 mM) has the potential to convert 2.86 g
CO2 to MgCO3 and CaCO3 per kg water (Figure 3). Thus, to sequester 10 Gt CO2 per year, around 3500 Gt of water would need to be processed annually, a quantity similar to our global annual level of water withdrawal (approximately 4000 Gt).86 On the other hand, 47 Gt of wastewater are processed annually87 in more than 14,700 treatment plants in the U.S.88 If a single CO2 abatement facility were to process 2000 Mt (i.e., the size of a large wastewater treatment plant)89 of seawater per year, then 1760 such plants would need to be built globally, with each plant being supplied with 10 Mt NaOH per year. Because the carbonate yield is limited by the content of divalent cations in the feed, enriching the Ca and Mg concentrations in the feed stream (e.g., using membranes that can selectively separate divalent cations, see Aguilar et al.90) would allow for the processing of a smaller quantity of water. Such pretreatment can obviously only be fulfilled while incurring a substantial energy penalty which appears unviable, in spite of the increase in the carbonate yields that would result.
Single-Step Carbon Sequestration and Storage (sCS2).
The energy consumption of electrochemically provoked mineralization-based CO2 management is associated primarily with the need to provide alkalinity to the process (see Methods and Figure 4). An ideal carbon sequestration process would not require consumable chemical inputs but would incur manu- facture, transport, handling, and storage costs. Ideally, the process could be powered using zero-carbon electrons, for example, from renewable sources. A conceptual chemical input- free single-step carbon sequestration and storage (sCS2) process is illustrated in Figures 5 and 6. Here, water (e.g., seawater) containing dissolved CO2in equilibrium with airflows through a porous conductive mesh/cathode. The application
of cathodic potentials leads to water’s electrolysis and locally elevated OH− concentrations at the mesh/water interface, which promotes the rapid combination of carbonate anions and metal cations while minimizing transport constraints and providing a substrate for heterogeneous nucleation (Figure 5). Specifically, flowing the electrolyte through the mesh’s pores minimizes the transport length scale (to the pore radius) for all ionic species (OH−, CO32−, and Me2+, where Me = Ca, Mg), while providing surface sites for the nucleation and growth of metal carbonates. This nature of process overcomes the kinetic barrier of carbonate precipitation in seawater in two ways. First,
heterogeneous nucleation lowers the induction time and the critical supersaturation for precipitation. Second, the persistence of a very high pH (>10−12) in the vicinity of the cathode results in Ω > 2000 (for pH 10; Figure 5c), depending on the overpotential, which significantly exceeds the critical Ω, thereby inducing immediate carbonate precipitation. It should be noted, however, that the nature of the precipitates (e.g., carbonates,
hydroxides, and mixtures thereof) that form is known to depend on the prevailing current density.91,92 Our own finite element analysis (FEA) indicates that electrolysis induces hyperalkaline conditions, rapidly, in proximity to the electroactive mesh (“electrode”) surface at reasonable overpotentials (≈0.5 V; Figure 5c). For example, the electrolyte volume within 200 μm
of the electrode/mesh surface (i.e., far larger than the pore size of the electroactive mesh envisioned for such applications) experiences hyperalkaline conditions within less than 1 s of the electrical polarization sufficient to induce carbonate precipitation to the bounding limits shown in Figure 3.
For reference herein, a 304 L stainless steel electrode is represented as a planar sheet, although a coarse mesh with an opening on the order of ∼20 μm is anticipated in practical applications. This analysis confirms that the application of mild potentials rapidly generates the needed alkalinity for carbonate
precipitation. Although this analysis neglects electromigration, electrophoresis, and/or gas evolution which can result in convective mixing, it offers an estimate of the overpotentials that are needed to induce rapid alkalinization and, in turn, carbonate precipitation. It should finally be noted that, in a symmetric electrolysis system, for example, where alkalinity is induced at the cathode, CO2 would be degassed at the anode because of the pH decrease (Figure 2a). An equivalent quantity of CO2, however, would be sorbed by the cathodic solution and then consumed rapidly to form carbonates. Although the
electrolysis of Cl− containing waters is challenging due to the
tendency to produce Cl2(g) under acidic conditions,30 this latter issue can be mitigated by using oxygen evolution reaction (OER)-selective coatings in the anode.95−97 Alternately, it would be necessary to apply commercial adsorption technolo- gies, for example, that utilize organic carbon-based solids (e.g.,coal, activated carbon) and/or zeolites to immobilize the Cl2(g) evolved and prevent its emission into the atmosphere.98,99
A significant advantage of mineralization using flow-through
electroactive mesh electrodes for the localized generation of alkalinity, in situ, is that it enhances the kinetics of precipitation (i.e., both nucleation and growth) because of the elevated pH,
supersaturation (Ω; Figures 3 and 5c),100 and temperature rise produced at the mesh surface induced by Joule heating (up to 60
°C, in solution).101 Obviously, the electrolytic nature of the
process requires conductive (e.g., metallic, or composite) meshes that are mechanically and chemically stable under cathodic conditions.102 Such mesh surfaces are expected to scale and foul during operation due to the formation of metal carbonates. Not only could the formation of carbonates result in substantive flux decay in the coarse-mesh architectures as considered herein, but the insulating nature of the mineral carbonates can also compromise the current density (i.e., thereby increasing the overpotential that is needed) and degrade energy efficiency. Therefore, reversing the applied potential, cyclically, to anodically generate O2 and H+ near the mesh surface is likely needed to remove deposits.103 However, anodic conditions can lead to rapid corrosion, particularly if Fe-based meshes are used. Therefore, we propose a method involving the continuous physical abrasion of deposited carbonates in a manner similar to that used to clean rotary drum filters (Figures 5b and 6). In these systems, the mesh’s surface is continuously scraped with a blade that disloges accumulated solids and re- exposes the mesh surface, simultaneously extracting the carbonate solids and regenerating the electrodes, although periodic polarity reversals may indeed continue to be required to achieve deep cleaning of strongly adhered carbonate precip- itates.
On the basis of our our data and observations, the carbonate solids that form present a wide dispersion of sizes and ordering (e.g., encompassing amorphous and crystalline variants). As such, in a realistic system, we expect a distribution of particles sizes where some form via heterogeneous processes and adhere to the electrode surface, and others form (also heterogeneously) but are either detached from the surface or form on alternate surfaces (e.g., dust particles) in the solution phase. However, dominantly, and because of the rapid decay in pH (i.e., corresponding to the driving force for carbonate formation) with increasing distance from the electrode surface (Figure 5c), it is likely that most of the precipitates will form at/in the vicinity of the electrode surfaces. Although colloidal-scale particles may indeed redeposit on the electrode surfaces depending on the flow conditions (i.e., Reynolds and Damkohler numbers), these particles could also remain suspended in solution and be rapidly carried away in the discharge stream. Although this would render direct discharge (e.g., in the seawater effluent) straightforward, it may raise other complexities, for example, difficulty to recover the particles if so desired. It is not clear, however, that this presents an issue vis-a-̀vis ocean discharge since the super- saturated conditions of oceanwater with respect to the mineral carbonates would ensure that these particles remain chemically stable (“undissolved”). However, these aspects require further evaluation using an operating system (i.e., at the bench and larger scales) not simply with regard to the need for electrode renewal and regeneration (e.g., by physical scraping or polarity reversals) but also with regard to the mode of discharge and/or recovery of the particles, for example, the need to flush the reactors with carbonate-supersaturated ocean waters, immedi- ately downstream of the cathode, to minimize the tendency for redissolution of the mineral precipitates. In any event, more generally, detailed analysis and demonstration, both at the bench and increasingly larger (pilot, commercial) scales would be needed to further evaluate and advance this concept. This includes (a) electrochemical screening of candidate electrode materials by measuring the kinetics (i.e., rate, yield, and
morphology) of carbonate precipitation as a function of the electrolytic conditions (e.g., applied potential, flow rates, current density), including single-pass removal levels of dissolved inorganic carbon, (b) fabrication and testing of flow reactors that integrate conductive mesh electrodes to enable rapid electrolytic carbonate formation in a controllable, modular manner, and (c) analysis of mass (water, carbon) and energy balances including influent and effluent compositions to confirm the energy intensity and demands of the process and the net carbon removal that is accomplished, including the effects on seawater chemistry, if a process of this manner were to be implemented and operated over the course of decades (or more) at global scales. Although unquestionably the sCS2 concept requires an energy input for carbon abatement, it also does the following: (a) It presents a lower energy demand than the vast majority of other direct air capture (DAC) approaches. (b) It allows straightfor- ward use of carbon-free electricity, for example, especially at times of excess. (c) It ensures end-to-end certainty around CO2 abatement by stabilizing mineral carbonates. In addition, rather than demanding the construction of new chloralkali plants, the electrolytic reactors envisaged herein can be modularly integrated with existing and future desalination plants thereby allowing CO2 removal and sequestration while producing potable water and hydrogen which can be used as a fuel.105 An added benefit of the sCS2 process is the production of softened water, an excellent feed for desalination plants. Currently, the energy cost of desalinating seawater can be estimated as 3.5 kWh per t of water, considering that seawater reverse osmosis (SWRO) requires 2−2.5 kWh per m3106 and the pretreatment steps, which is water softening, consume 0.3−1.0 kWh per m3 of seawater.107 Combining CO2 mineralization-based pretreatment and desalination can lead to an energy use that is 9% lower than the total energy consumption of the two processes, operating separately. Nonetheless, a massive buildout of plants would be needed, globally, incurring trillions of dollars of expenditure to achieve the required water processing throughputs and carbon mitigation scale that is required to limit global warming to the 1.5 °C threshold.46
The energy requirements of electrolytic mineralization-based CO2 removal are formidable. For example, mineralizing 660 t of CO2 per h would require 1500 MW of power. It would be necessary to buildout 1760 plants at this scale, around the world, each having 8410 mesh-electrode units, to annually mineralize 10 Gt of CO2, while consuming more than 20 PWh of electricity. Today, the world’s largest solar power plant located in India produces 2200 MW and occupies 40 km2.108 The capital cost for installation of solar power plants is around $1 million per 1 MW.109 Thus, about $1.4 trillion (around 7% of the U.S. GDP or 2% of global GDP; Note: for context, the 2020 U.S. COVID-19 stimulus bill amounted to $2 trillion)110,111 would be needed for emplacing solar plants that would power sCS2-based CO2 removal. In general, however, this effort and many other NETs will be promoted by the ongoing pace of energy transitions and the reduction in the price of renewable energy. For example, the U.S. Department of Energy’s SunShot program aims to achieve photoelectric generation at $30 per MWh by 2030.112 In April 2020, the California Independent System Operator (CAISO) lists prices of around $35 per MWh for renewable energy.113 This is reflective of a power excess market that exists in some jurisdictions (e.g., California, Texas, etc.), where excess renewable electricity could be directed to CO2 removal where incentivized. For reference, the total installed
photovoltaic capacity globally was around 500 GW in 2018 and is expected to double to 1 TW in 2022.114,115 A previous estimate has shown that based on the availability of renewable energy and process efficiencies, the CO2 removal potential that is associated with electrolytic H2 generation is from 90 to 900 Gt per year.116 This is an important outcome, albeit one that needs to further accelerate in the years to come.
On the basis of current forecasts, zero-carbon renewable energy is expected to only encompass two-thirds of the energy mix, globally, by 2040.117 However, we postulate that depending on the energy intensity of proposed NETs, even fossil fuels could be used to power CO2 abatement solutions. For example, natural gas combustion (NGC), assuming current industrial efficiencies, results in the release of 1 t of CO2 per 2 MWh of electricity generation, which yields a specific intensity ratio (SIR) of 0.5 t of CO2 emitted/MWh of energy produced. As such, an end-to-end carbon abatement process (i.e., which involves CO2 immobiliza- tion, ad infinitum, and not simply carbon capture) which consumes less than 2 MWh per tonne of CO2 abated could be considered a true NET if powered by NGC. Obviously, if natural gas were to be used to power NETs, it is necessary that the energy intensity of the NET should be far smaller than 2 MWh of energy consumed per t of CO2 abated to meaningfully reduce atmospheric CO2 accumulation, while ensuring its (net) removal. On a grid-wide basis (U.S. average emissions factor of 743 g/kWh of electricity) and setting solar electricity (emissions factor of 25 g/kWh of electricity)3 as a baseline zero-carbon electricity reference, it is important to ask whether renewable energy should be used to (1) displace fossil fuel-based power generation or (2) used to power NETs. The answer is complex, but in short, it depends on the energy intensity of the CO2 removal process. For the case of NGC-based electricity generation, in general, a NET needs to feature an energy intensity of no more than 1.34 MWh per t of CO2 removed. The conclusion is that the sCS2 process as outlined herein, if deployed in a format where the energy in the cogenerated H2(g)
can be recovered (0.8−1.2 MWh per t of CO2 removed), features an energy intensity that is sufficiently low to be worthy
of deployment as a renewable energy powered NET, rather than displacing sources of NGC-powered electricity generation by renewable energy generation.
Implementation and Effects on Ocean Chemistry. The
reduction in pH resulting from alkaline cation depletion, if carried out in an uncontrolled manner, could decrease the carbon storage capacity of seawater by 16 Gt CO2 (2.8 × 10−7 mol CO2 per kg water) on an annual basis (see Methods); i.e., a net accumulation of 6 Gt of CO2 would result in the atmosphere. Although this analysis is rather simplified and conservative and
better consideration of oceanic circulation and cation replenish- ment fluxes is required, we nevertheless seek to assess the net effects of such perturbations. Thus, to prevent any pH decrease of seawater, it is proposed to discharge the sCS2 process effluent that is depleted of cations in regions of robust near-coastal mixing to allow rapid equilibration of cation concentrations with existing seawater. The other possibility given the replenishment of oceanic alkaline cation levels via riverine discharge implies the siting of seawater mineralization plants in the vicinity of a delta which would ensure replenishment of cation concentrationsin the effluentvia riverine-sourced alkalinity (i.e., Ca and Mg species).
Significantly, however, even if cation replenishment is
required, it can also be readily achieved in an electrochemical system by exploting the acidity generation that is consequent at
the anode. Specifically, in an engineered system, electrolytic (re)alkalinization of the reject seawater stream can be performed by the acidity-catalyzed dissolution of mafic and ultramafic rocks and to a lesser extent by industrial solids including coal combustion and metal processing residues, in the style of enhanced weathering,118 using the acidity cogenerated in the sCS2 process or perhaps, by the use of ion-exchange systems that are common to water treatment applications.
More broadly, this analysis shows that a unit of alkalinity stores a greater amount of CO2 in an aqueous form than as a solid. Although 2 mol of OH− are required for each mole of C stored as a carbonate solid, only 1.2 mol of OH− are required per mole of C stored as dissolved ions.9 As a result, increasing the pH from 8 to 9 (i.e., 1 to 10 μM OH−) solubilizes an additional 33 mmol CO2 per kg water (Figure 1). This indicates that, expectedly, ocean acidification can be countered either by
increasing the seawater alkalinity or by the removal of dissolved CO2 (in seawater). Although we have focused on the latter strategy, we recognize that any strategy that removes CO2 from the atmosphere should be carefully combined with seawater
(re)alkalinization to enhance its CO2 capacity as driven by the ocean−atmosphere equilibrium119 to prevent CO2 from degassing back to the atmosphere (as atmospheric CO2 concentrations are lowered, in time). Hence, “synthetic alkalinity matching” could be performed to the extent that the pH of the discharge stream is either the same as the input seawater to minimize perturbations in ocean chemistry or the
discharge water can be designed to have a greater pH (i.e., higher [Ca] and/or [Mg]) than the extracted seawater to render a further enhanced carbon abatement benefit.9 The latter strategy would require a detailed understanding of the near- and long- term effects on ocean chemistry prior to implementation. In any event, we specifically promote the immobilization of CO2 however, within a solid and not in an aqueous form (e.g., dissolved carbonate and bicarbonate species)because it simplifies the logistics associated with carbon storage (i.e., into a solid waste handling problem) and also ensures indefinite storage of CO2 in a chemically stabilized form while eliminating all need for monitoring and verification of sequestered CO2. This approach not only avoids a need to actively induce changes in the ocean chemistry but also eliminates any risk of CO2 degassing that may come about with ongoing climate change and a rise in the ocean surface temperature. For example, a 1 °C increase in the average ocean temperature (e.g., 20th century
average ocean surface temperature is 16 °C120) would result in a reduction in CO2 solubility by ∼7 × 10−6 mol CO2/kg water, which is a decrease in the total oceanic CO2 storage capacity by 400 Gt.
Fate of Carbonate Solids. Assuming stoichiometry and the precipitation of calcite, magnesite, hydroxy-carbonates, etc., the removal of at least 10 Gt of CO2 from the atmosphere (i.e, dissolved in seawater) will result in the production of over 20 Gt of solids annually. Some of these solids could substitute the global mineral carbonate market, which amounts to ∼10−20 Gt
per year,121 spanning construction materials (aggregates) and specialty applications. In the U.S., 68% of produced crushed stone is composed of carbonate rocks, about 1 Gt of production of which is used for construction and as raw material for cement production.122 Net negative emissions are immediately realized when the carbonates are used as aggregates or the limestone as cement kiln feed; the latter in effect reduces the carbon intensity
of cement production by ∼60%. The solids that cannot be
utilized can be disposed of via existing solid waste management
strategies. In 2016, global municipal solid waste generation and industrial, agricultural, construction, and demolition waste amounted to about 25 Gt.123,124 Landfilling of solid waste costs about $45 per t in the U.S.,125 and landfill disposal of 10 Gt of carbonate solids will require about 6.8 km3 (6.8 billion m3) of space per year. Rather than building new landfills, the solids can be stored at defunct mines. In 2017, we extracted 53 Gt of metal and nonmetallic ore material, 15 Gt of fossil fuels, and 24 Gt of biomass worldwide.126,127 However, offsite storage would require transportation of the solids, costing about $0.03 per m3 per km, a substantial expense, especially if the solids needed to be transported over long distances.128 More realistically, if using seawater as the alkaline source, the precipitates could be redeposited in the oceans (i.e., in the style of desalination brines, where since the oceans are oversaturated with respect to calcite and magnesite, these solids will remain stable and unreactive; Figure 6) or used for land reclamation and erosion prevention purposes in close vicinity to the site of production.
Under the London Protocol as amended and enforced in 2006, marine dumping is prohibited except for possibly acceptable wastes as outlined in the “reverse list” of Annex 1.129 Calcium and magnesium carbonates may qualify as “inert,
geological materials” which are permissible for disposal in the ocean where they will remain stable since near-surface seawater is supersaturated with respect to both phases.130,131 If dissolved Ca and Mg are taken from sources other than seawater (e.g., saline groundwater), the precipitated Ca and Mg carbonates can be used to buffer decreasing ocean pH caused by atmospheric
CO2 absorption by the addition and dissolution of lime- stone.132,133
Coming back to land reclamation, a simple model of shoreline migration in Southern California approximates a recession of about 30 m for 1 m of sea level rise.134 Assuming that the continental shelf has an average depth of 50 m, generation of 20 Gt of solids can reverse this effect over a shoreline extending 4500 km, around half the length of Florida’s gulf coast.135 Creation of new land by CO2 mineralization-derived solids may not only address future CO2 emissions but could also potentially reverse one of the most prominent effects of climate change. The crisis of disappearing landmass and habitat by sea level rise can be addressed, while providing a CO2 storage solution that is both permanent and does not require continuous monitoring. An in- depth analysis of the mechanisms for subsidizing CO2 management via the sCS2 approach, particularly the associated capital cost, is beyond the scope of this work. Nonetheless, the recent 45Q136 tax credit in the U.S. and California’s low-carbon fuel standard (LCFS)137 incentivize carbon mitigation by
implicitly pricing CO2 between $35−$180 per t. Such incentives
offer new pathways for enabling, empowering, and accelerating global-scale CO2 mitigation and reduction.
SUMMARY AND CONCLUSIONS
The proposed sCS2 approach offers a route to accelerate carbon management in a cost-effective manner without being hostage to the creation and implementation of supportive policy and associated derisking (e.g., via Sovereign guarantees) since the mineralized CO2 that is formed (“solid carbonates”) can be benignly disposed of at the Earth’s surface. The large and durable carbon storage capacity, the benign nature of the process, and the elimination of the risk of CO2 release ensure the viability of the process for long-term Gt-scale CO2 waste management. Thus, we submit this sCS2 approach as another possible pathway to augment our growing and increasingly
important technological portfolio of CO2 mitigation and management solutions. Obviously, a major gap in the supply of low-carbon and carbon-free electricity needs to be closed for this to achieve widespread viability (Note: This is true, broadly speaking, for all “negative emissions technologies”, today). However, given that carbon storage needs to last for thousands of years, the combination of electrolytic seawater CO2 mineralization in combination with accelerated silicate/ carbonate weathering offers us a viable, environmentally benign, and potentially more acceptable approach toward solving the global carbon crisis than traditional geological sequestration, especially as we seek to accelerate and deploy (i.e., scaleup and scaleout) NETs in the short-to-medium term in the next decade or so.
BROADER CONTEXT
Over the upcoming years, unprecedented reductions in CO2 emissions and atmospheric CO2 content are necessary to limit and mitigate ongoing climate change. The underground injection of CO2 captured from point sources remains our leading candidate for global carbon management along with supplementary approaches conditioned on land use change(s) and the chemical conversion (i.e., often utilization) of CO2.8,138 However, the risk of CO2 migration and leakage appears to handicap the acceptability of geological carbon capture and sequestration, and significant advancements need to be realized to ensure durable carbon storage at the phenomenal rates that are needed to mitigate climate change. The sCS2 concept rationalized in this perspective has the potential to effect the removal of CO2, at a gigatonne(s) scale annually, while utilizing alkalinity generated electrolytically in such a way that the process could be operated either wholly, or partially, using renewable energy.
AUTHOR INFORMATION
Corresponding Author
Gaurav N. Sant − Department of Civil and Environmental Engineering, Institute for Carbon Management, Department of Materials Science and Engineering, and California Nanosystems Institute, University of California, Los Angeles, California 90095, United States; —orcid.org/0000-0002- —1124-5498; Phone: (310) 206-3084; Email: [email protected]
Authors
Erika Callagon La Plante − Department of Civil and Environmental Engineering and Institute for Carbon Management, University of California, Los Angeles, California 90095, United States; Department of Materials Science and Engineering, University of Texas at Arlington, Arlington, Texas 76019, United States; 112222orcid.org/0000-0002-5273-9523
Dante A. Simonetti − Institute for Carbon Management and Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, California 90095, United States; 11222orcid.org/0000-0002-5708-460X
Jingbo Wang − Department of Civil and Environmental Engineering, University of California, Los Angeles, California 90095, United States
Abdulaziz Al-Turki − Department of Chemical and Biomolecular Engineering, University of California, Los Angeles, California 90095, United States
Xin Chen − Department of Civil and Environmental Engineering and Institute for Carbon Management, University
of California, Los Angeles, California 90095, United States; 11222orcid.org/0000-0003-3273-4431
David Jassby − Department of Civil and Environmental Engineering and Institute for Carbon Management, University of California, Los Angeles, California 90095, United States;
—orcid.org/0000-0002-2133-2536
Complete contact information is available at: .
https://pubs.acs.org/10.1021/acssuschemeng.0c08561
Notes The authors declare no competing financial interest.
Biographies

Erika Callagon La Plante is an assistant professor in the Department of Materials Science and Engineering and the Center for Advanced Construction Materials at the University of Texas at Arlington. Her research expertise is in the area of materials chemistry, mineral−fluid interfacial structures and reactions, and low-temperature geochemistry. She previously held appointments at the Department of Civil and Environmental Engineering and Institute for Carbon Management at the University of California, Los Angeles (UCLA) initially as a postdoctoral researcher and thereafter as an assistant project scientist and lecturer from 2016 to 2020. Erika received her B.S. in Geology from the University of the Philippines in 2010 and her Ph.D. in Geochemistry from the Department of Earth and Environmental Sciences at the University of Illinois at Chicago in 2016.

Dante A. Simonetti is an assistant professor in the Department of Chemical and Biomolecular Engineering and Institute for Carbon Management at the University of California, Los Angeles (UCLA). His research expertise is in the area of catalytic reaction synthesis, adsorptive separation processes, and process integration and intensification, and he holds multiple patents in these areas. Dante’s research has been recognized by awards such as the Hellmann Fellowship and the American Chemical Society’s New Investigator Award. Prior to UCLA, Dante was a scientific staff member at UOP- Honeywell where he was a project leader for R&D activities related to natural gas processing and oil and gas refining applications. Dante received his B.S. from the University of Notre Dame in 2003 and his Ph.D. from the University of Wisconsin−Madison in 2008, both in chemical engineering, and he completed postdoctoral training in chemical and biomolecular engineering at the University of California, Berkeley (UCB).

Jingbo Wang is a postdoctoral scholar in the Department of Civil and Environmental Engineering at the University of California, Los Angeles (UCLA). Her research interests include water treatment and desalination and the application of nanotechnology to membrane separation processes for water, energy, and resource recovery. Jingbo received her B. Eng. from Wuhan University in 2012 and her Ph.D. in environmental sciences and engineering from the University of North Carolina at Chapel Hill in 2018.

Abdulaziz Al-Turki is an assistant professor at King Abdulaziz University in Jeddah, Saudi Arabia. Prior to UCLA, Abdulaziz was a Ph.D. student in the Department of Chemical and Biomolecular Engineering at UCLA, and before then, he was a process/supply planning engineer with Saudi Aramco. Abdulaziz’s research expertise is in modeling and simulation and intensification of chemical engineering processes. Abdulaziz received his B.S. in chemical engineering from the University of Leeds, U.K., in 2012 and his Ph.D. in chemical and biomolecular engineering from UCLA in 2020.

Xin Chen is an associate development engineer in the Department of Civil and Environmental Engineering and the Institute for Carbon Management at the University of California, Los Angeles (UCLA). Xin’s research expertise is in the area of electrochemical reactions related to the formation, alteration, and corrosion of structural materials with applications in nuclear power generation, clean energy, and environmental technologies, and biomedical systems. Xin received his

B.S. in materials science and engineering from the Beijing Institute of Technology in 2011 and his M.S. and Ph.D. degrees in materials science and materials engineering from the Illinois Institute of Technology and the University of Illinois at Chicago in 2014 and 2017, respectively.

David Jassby is an associate professor in the Department of Civil and Environmental Engineering and Institute for Carbon Management at the University of California, Los Angeles (UCLA). Before UCLA, he was an assistant professor in the Department of Chemical and Environmental Engineering at the University of California, Riverside. David’s research expertise is in the domain of water treatment technologies, environmental applications of nanotechnology, and environmental electrochemistry with applications related to industrial wastewater treatment, oil/water separations, desalination, and the electrochemical treatment of groundwater. David received his B.Sc. in biology from Hebrew University in 2002, an M.S. in civil and environmental engineering from UC−Davis in 2005, and his Ph.D. in civil and environmental engineering from Duke University in 2011.
Gaurav N. Sant is a professor and a Samueli Fellow in the Departments of Civil and Environmental Engineering and Materials Science and Engineering and the California Nanosystems Institute at the University of California, Los Angeles (UCLA) where he is also the director of UCLA’s Institute for Carbon Management. Gaurav’s research expertise encompasses affecting and quantifying aqueous reactions at surfaces and interfaces including dissolution, precipitation, and corrosion with applications to construction materials, clean energy, and environmental technologies and decarbonizing heavy industry operations. Gaurav’s research has been recognized by awards including the National Science Foundation’s CAREER Award, a Hellmann Fellowship, RILEM’s Gustavo Collonnetti Medal, and the American Concrete Institute’s Walter P. Murphy Jr., and J.C Roumain Awards. Gaurav received his B.S., M.S., and Ph.D. in civil engineering from Purdue University in 2006, 2007, and 2009, respectively, and completed post-doctoral training in materials science at the Ecole Polytechnique Federale de Lausanne (EPFL) in Lausanne, Switzerland.
ACKNOWLEDGMENTS
The authors acknowledge financial support for this research
provisioned by the U.S. Department of Energy (DE-FE0029825, DE-FE0031718, DE-FE0031705), The Anthony and Jeanne Pritzker Family Foundation, The Grantham Foundation for the Protection of the Environment, The National Science Foundation (DMREF: 1922167), the U.S.-China Clean Energy Research Center for Water-Energy Technologies (CERC- WET), the University of Texas, Arlington, and UCLA’s Institute for Carbon Management. The contents of this prespective reflect the views and opinions of the authors, who are responsible for the accuracy of the data sets presented herein, and do not reflect the views and/or policies of the funding agency, nor do the contents constitute a specification, standard, or regulation. The authors acknowledge James McDermott and Benjamin Stone (1PointFive Inc. and Rusheen Capital Manage- ment) for their assistance in the analysis of capital costs and the levelized cost of carbon abatement (LCCA), Edward Muller (Transocean/Aerovironment) and Anthony Ku (NiCE Amer- ica) for discussions regarding clean energy/hydrogen economy, and Raghubir Gupta (Susteon Inc.) for technology and cost- mapping of alkaline electrolyzers.
REFERENCES
• United Nations Environment Programme. Emissions Gap Report 2019; United Nations Environment Programm: Nairobi, 2019.
• Core Writing Team. Pachauri, R. K.; Meyer, L. A. Climate Change 2014: Synthesis Report. Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change; IPCC: Geneva, Switzerland, 2014.
• National Academies of Sciences, Engineering, and Medicine. Negative Emissions Technologies and Reliable Sequestration: A Research Agenda; National Academies Press, 2019.
• National Energy Technology Laboratory, U.S. Department of Energy. Carbon Storage Atlas, Fifth ed.; U.S. Department of Energy, 2015.
• Jahediesfanjani, H.; Warwick, P. D.; Anderson, S. T. …Estimating
the Pressure-Limited CO2 Injection and Storage Capacity of the United …States Saline Formations: Effect of the Presence of Hydrocarbon …Reservoirs. Int. J. Greenhouse Gas Control 2018, 79, 14−24.
• United States Environmental Protection Agency. Geologic CO2
Sequestration Technology and Cost Analysis; Technical Support Docu- ment; United States Environmental Protection Agency, 2008.
• Gattuso, J.-P.; Magnan, A. K.; Bopp, L.; Cheung, W. W. L.; Duarte,
C. M.; Hinkel, J.; Mcleod, E.; Micheli, F.; Oschlies, A.; Williamson, P.; Bille,́R.; Chalastani, V. I.; Gates, R. D.; Irisson, J.-O.; Middelburg, J. J.; Pörtner, H.-O.; Rau, G. H. ..Ocean Solutions to Address Climate Change ..and Its Effects on Marine Ecosystems. Front. Mar. Sci. 2018, 5, 1−18.
• National Academies of Sciences, Engineering, and Medicine.
Gaseous Carbon Waste Streams Utilization: Status and Research Needs; The National Academies Press: Washington, DC, 2019.
• Renforth, P.; Henderson, G. ./Assessing Ocean Alkalinity for ./Carbon Sequestration. Rev. Geophys. 2017, 55 (3), 636−674.
• Millero, F. J.; Feistel, R.; Wright, D. G.; McDougall, T. J. …The
Composition of Standard Seawater and the Definition of the Reference- …Composition Salinity Scale. Deep Sea Res., Part I 2008, 55 (1), 50−72.
• Matter, J. M.; Stute, M.; Snaebjornsdottir, S. O.; Oelkers, E. H.;
Gislason, S. R.; Aradottir, E. S.; Sigfusson, B.; Gunnarsson, I.; Sigurdardottir, H.; Gunnlaugsson, E.; Axelsson, G.; Alfredsson, H. A.; Wolff-Boenisch, D.; Mesfin, K.; Taya, D. F. d. l. R.; Hall, J.; Dideriksen, K.; Broecker, W. S. .Rapid Carbon Mineralization for Permanent .Disposal of Anthropogenic Carbon Dioxide Emissions. Science 2016,
352 (6291), 1312−1314.
• Zevenhoven, R.; Eloneva, S.; Teir, S. …Chemical Fixation of CO2 in …Carbonates: Routes to Valuable Products and Long-Term Storage. Catal. Today 2006, 115 (1), 73−79.
• Chang, R.; Kim, S.; Lee, S.; Choi, S.; Kim, M.; Park, Y. ..Calcium
Carbonate Precipitation for CO2 Storage and Utilization: A Review of ..the Carbonate Crystallization and Polymorphism. Front. Energy Res. 2017, 5, 1−12.
• Berger, G.; Cadore, E.; Schott, J.; Dove, P. M. ./-()-Dissolution Rate of
Quartz in Lead and Sodium Electrolyte Solutions between 25 and ./-()-300°C: Effect of the Nature of Surface Complexes and Reaction ./-()-Affinity. Geochim. Cosmochim. Acta 1994, 58 (2), 541−551.
• Dove, P. M. .-()-The Dissolution Kinetics of Quartz in Aqueous .-()-Mixed Cation Solutions. Geochim. Cosmochim. Acta 1999, 63 (22),
3715−3727.
• Oey, T.; Kumar, A.; Pignatelli, I.; Yu, Y.; Neithalath, N.; Bullard,
J. W.; Bauchy, M.; Sant, G. .Topological Controls on the Dissolution .Kinetics of Glassy Aluminosilicates. J. Am. Ceram. Soc. 2017, 100 (12), 5521−5527.
• Hamilton, J. P.; Brantley, S. L.; Pantano, C. G.; Criscenti, L. J.;
Kubicki, J. D. .-()-Dissolution of Nepheline, Jadeite and Albite Glasses: .-()-Toward Better Models for Aluminosilicate Dissolution. Geochim. Cosmochim. Acta 2001, 65 (21), 3683−3702.
• Said, A.; Mattila, O.; Eloneva, S.; Jar̈vinen, M. …Enhancement of
Calcium Dissolution from Steel Slag by Ultrasound. Chem. Eng. Process.
2015, 89, 1−8.
• Liu, Q.; Maroto-Valer, M. M. …Studies of PH Buffer Systems to …Promote Carbonate Formation for CO2 Sequestration in Brines. Fuel Process. Technol. 2012, 98, 6−13.
• Constantz, B. R.; Schneider, J.; Bewernitz, M. Ammonia
Mediated Carbon Dioxide (CO2) Sequestration Methods and Systems.
U.S. Patent US20170274318 A1, September 28, 2017.
• Imbabi, M. S.-E.; Glasser, F. Carbon Dioxide Capture and Conversion Methods and Systems. U.S. Patent US20190232216 A1, August 1, 2019.
• Jones, J. D. Removing Carbon Dioxide from Waste Streams through Co-Generation of Carbonate and/or Bicarbonate Minerals.
U.S. Patent US8741244 B2, June 3, 2014.
• Constantz, B. R.; Youngs, A.; Holland, T. C. CO2-Sequestering Formed Building Materials. World Patent WO2010039903 A1, April 8, 2010.
• Esposito, D. V. …Membraneless Electrolyzers for Low-Cost …Hydrogen Production in a Renewable Energy Future. Joule 2017, 1 (4), 651−658.
• Talabi, O. O.; Dorfi, A. E.; O’Neil, G. D.; Esposito, D. V.
Membraneless Electrolyzers for the Simultaneous Production of Acid .7and Base. Chem. Commun. 2017, 53 (57), 8006−8009.
• Eisaman, M. D.; Parajuly, K.; Tuganov, A.; Eldershaw, C.; Chang, N.; Littau, K. A. .2CO2 Extraction from Seawater Using Bipolar .2Membrane Electrodialysis. Energy Environ. Sci. 2012, 5 (6), 7346−
7352.
• de Lannoy, C.-F.; Eisaman, M. D.; Jose, A.; Karnitz, S. D.; DeVaul, R. W.; Hannun, K.; Rivest, J. L. B. …Indirect Ocean Capture of …Atmospheric CO2: Part I. Prototype of a Negative Emissions …Technology. Int. J. Greenhouse Gas Control 2018, 70, 243−253.
• Lu, L.; Huang, Z.; Rau, G. H.; Ren, Z. J. .5Microbial Electrolytic .5Carbon Capture for Carbon Negative and Energy Positive Wastewater .5Treatment. Environ. Sci. Technol. 2015, 49 (13), 8193−8201.
• House, K. Z.; House, C. H.; Schrag, D. P.; Aziz, M. J. .Electrochemical Acceleration of Chemical Weathering as an Energeti- .cally Feasible Approach to Mitigating Anthropogenic Climate Change. Environ. Sci. Technol. 2007, 41 (24), 8464−8470.
• Rau, G. H.; Carroll, S. A.; Bourcier, W. L.; Singleton, M. J.; Smith,
M. M.; Aines, R. D. .Direct Electrolytic Dissolution of Silicate Minerals .for Air CO2 Mitigation and Carbon-Negative H2 Production. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (25), 10095−10100.
• Plummer, L. N.; Busenberg, E. ./-()-The Solubilities of Calcite, ./-()-Aragonite and Vaterite in CO2-H2O Solutions between 0 and 90°C, and ./-()-an Evaluation of the Aqueous Model for the System CaCO3-CO2-H2O. Geochim. Cosmochim. Acta 1982, 46 (6), 1011−1040.
• Andersson, M. P.; Rodriguez-Blanco, J. D.; Stipp, S. L. S. …Is
Bicarbonate Stable in and on the Calcite Surface? Geochim. Cosmochim. Acta 2016, 176, 198−205.
• Power, I. M.; Kenward, P. A.; Dipple, G. M.; Raudsepp, M. .7Room .7Temperature Magnesite Precipitation. Cryst. Growth Des. 2017, 17 (11), 5652−5659.
• Roberts, J. A.; Kenward, P. A.; Fowle, D. A.; Goldstein, R. H.; Gonzaĺez, L. A.; Moore, D. S. .Surface Chemistry Allows for Abiotic .Precipitation of Dolomite at Low Temperature. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (36), 14540−14545.
• Millero, F. J.; Roy, R. N. A Chemical Equilibrium Model for the Carbonate System in Natural Waters. Croat. Chem. Acta 1997, 70 (1), 1−38.
• Millero, F. J.; Graham, T. B.; Huang, F.; Bustos-Serrano, H.; Pierrot, D. …Dissociation Constants of Carbonic Acid in Seawater as a …Function of Salinity and Temperature. Mar. Chem. 2006, 100 (1), 80− 94.
• Mitchell, M. J.; Jensen, O. E.; Cliffe, K. A.; Maroto-Valer, M. M. ..A ..Model of Carbon Dioxide Dissolution and Mineral Carbonation ..Kinetics. Proc. R. Soc. London, Ser. A 2010, 466 (2117), 1265−1290.
• Stumm, W.; Morgan, J. J. Aquatic Chemistry: Chemical Equilibria and Rates in Natural Waters; John Wiley & Sons, 2012; Vol. 126.
• Wang, X.; Conway, W.; Burns, R.; McCann, N.; Maeder, M. .Comprehensive Study of the Hydration and Dehydration Reactions of .Carbon Dioxide in Aqueous Solution. J. Phys. Chem. A 2010, 114 (4), 1734−1740.
• Bustillos, S.; Alturki, A.; Prentice, D.; La Plante, E. C.; Rogers, M.; Keller, M.; Ragipani, R.; Wang, B.; Sant, G.; Simonetti, D. A. ..Implementation of Ion Exchange Processes for Carbon Dioxide ..Mineralization Using Industrial Waste Streams. Front. Energy Res.
2020, 8, 1−17.
• Nancollas, G. H.; Reddy, M. M. ./-()-The Crystallization of Calcium ./-()-Carbonate. II. Calcite Growth Mechanism. J. Colloid Interface Sci. 1971, 37 (4), 824−830.
• U.S. Department of Energy, Office of Energy Efficiency and Renewable Energy. Bandwidth Study on Energy Use and Potential Energy Savings Opportunities in U.S. Seawater Desalination Systems; U.S. Department of Energy, 2017.
• WateReuse Association Desalination Committee. Seawater Desalination Power Consumption; White Paper; WateReuseDesalina- tionCommittee, 2011.
• Plappally, A. K.; Lienhard, V. J. H. …Energy Requirements for …Water Production, Treatment, End Use, Reclamation, and Disposal. Renewable Sustainable Energy Rev. 2012, 16 (7), 4818−4848.
• Eisaman, M. D. …Negative Emissions Technologies: The …Tradeoffs of Air-Capture Economics. Joule 2020, 4 (3), 516−520.
• Eisaman, M. D.; Rivest, J. L. B.; Karnitz, S. D.; de Lannoy, C.-F.; Jose, A.; DeVaul, R. W.; Hannun, K. …Indirect Ocean Capture of …Atmospheric CO2: Part II. Understanding the Cost of Negative …Emissions. Int. J. Greenhouse Gas Control 2018, 70, 254−261.
• U.S. Department of Energy, Office of Energy Efficiency and
Renewable Energy. Industrial Technologies Program: Advanced Chlor- Alkali Technology; U.S. Department of Energy, 2006.
• U.S. Energy Information Administration. Electricity Explained: Factors Affecting Electricity Prices.
eia.gov/energyexplained/index.php?page=electricity_factors_
affecting_prices
(accessed Jun 3, 2019).
• Jones, J.; Barton, C.; Clayton, M.; Yablonsky, A.; Legere, D. Sky Mine Carbon Mineralization Pilot Project; Skyonic Corporation, 2010. ./DOI: 10.2172/1027801.
• Renforth, P.; Washbourne, C.-L.; Taylder, J.; Manning, D. A. C. .Silicate Production and Availability for Mineral Carbonation. Environ. Sci. Technol. 2011, 45 (6), 2035−2041.
• Thiel, G. P.; Kumar, A.; Goḿez-Gonzaĺez, A.; Lienhard, J. H.
Utilization of Desalination Brine for Sodium Hydroxide Production: .7Technologies, Engineering Principles, Recovery Limits, and Future .7Directions. ACS Sustainable Chem. Eng. 2017, 5 (12), 11147−11162.
• Kheshgi, H. S. ./-()Sequestering Atmospheric Carbon Dioxide by ./-()Increasing Ocean Alkalinity. Energy 1995, 20 (9), 915−922.
• Rau, G. H. .Electrochemical Splitting of Calcium Carbonate to .Increase Solution Alkalinity: Implications for Mitigation of Carbon .Dioxide and Ocean Acidity. Environ. Sci. Technol. 2008, 42 (23), 8935− 8940.
• Aspen Technology, Inc. Aspen Plus.
• Chen, C.-C.; Song, Y. .Generalized Electrolyte-NRTL Model for .Mixed-Solvent Electrolyte Systems. AIChE J. 2004, 50 (8), 1928−1941.
• Alturki, A. A. Integration of Sorption Enhanced Operations into Sustainable Processes; Thesis, University of California: Los Angeles, 2020.
• Bui, M.; Tait, P.; Lucquiaud, M.; Mac Dowell, N. …Dynamic …Operation and Modelling of Amine-Based CO2 Capture at Pilot Scale. Int. J. Greenhouse Gas Control 2018, 79, 134−153.
• Mota-Martinez, M. T.; Brandl, P.; Hallett, J. P.; Mac Dowell, N. .8Challenges and Opportunities for the Utilisation of Ionic Liquids as .8Solvents for CO2 Capture. Mol. Syst. Des. Eng. 2018, 3 (3), 560−571.
• Cui, Q.; Lu, H.; Li, C.; Singh, S.; Ba, L.; Zhao, X.; Ku, A. Y. …China
Baseline Coal-Fired Power Plant with Post-Combustion CO2 Capture:
1. Definitions and Performance. Int. J. Greenhouse Gas Control 2018, 78, 37−47.
• Sakwattanapong, R.; Aroonwilas, A.; Veawab, A. .Behavior of .Reboiler Heat Duty for CO2 Capture Plants Using Regenerable Single .and Blended Alkanolamines. Ind. Eng. Chem. Res. 2005, 44 (12), 4465− 4473.
• Arshad, M. W.; Fosbøl, P. L.; von Solms, N.; Svendsen, H. F.; Thomsen, K. …Equilibrium Solubility of CO2 in Alkanolamines. Energy Procedia 2014, 51, 217−223.
• Ivy, J. Summary of Electrolytic Hydrogen Production; Milestone Completion Report NREL/MP-560-36734; National Renewable Energy Laboratory, 2004.
• The National Academies of Sciences. Our Energy Sources. Fossil Fuels. Natural Gas.
http://needtoknow.nas.edu/energy/energy- sources/fossil-fuels/natural-gas/ (accessed Apr 6, 2020).
• Karimi, M.; Hillestad, M.; Svendsen, H. F. .Natural Gas .Combined Cycle Power Plant Integrated to Capture Plant. Energy Fuels 2012, 26 (3), 1805−1813.
• Eichman, J.; Townsend, A.; Melaina, M. Economic Assessment of Hydrogen Technologies Participating in California Electricity Markets; NREL/TP-5400-65856; National Renewable Energy Laboratory, 2016.
• U.S. Environmental Protection Agency, Combined Heat and Power Partnership. Catalog of CHP Technologies: Section 6. Technology Characterization – Fuel Cells, March 2015. -6
epa.gov/sites/-6production/files/2015-07/documents/catalog_of_chp_technologies_
-6section_6._technology_characterization_-_fuel_cells.pdf (accessed January 2021).
• Department of Energy, Office of Energy Efficiency & Renewable
Energy. DOE Technical Targets for Hydrogen Production from Electrolysis.
https://www.energy.gov/eere/fuelcells/doe-technical- targets-hydrogen-production-electrolysis (accessed January 2021).
• Ciampoli, P. LADWP Board OKs PPAs for Massive Solar plus Storage Project; American Public Power Association, September 11, 2019.
• McDuff, R. E.; Morel, F. M. M. .aThe Geochemical Control of .aSeawater (Sillen Revisited). Environ. Sci. Technol. 1980, 14 (10), 1182− 1186.
• Shaojun, Z.; Mucci, A. ./-()Calcite Precipitation in Seawater Using a ./-()Constant Addition Technique: A New Overall Reaction Kinetic ./-()Expression. Geochim. Cosmochim. Acta 1993, 57 (7), 1409−1417.
• Lin, Y.-P.; Singer, P. C.; Aiken, G. R. .Inhibition of Calcite
Precipitation by Natural Organic Material: Kinetics, Mechanism, and .Thermodynamics. Environ. Sci. Technol. 2005, 39 (17), 6420−6428.
• Sabbides, T.; Giannimaras, E.; Koutsoukos, P. G. ./The ./Precipitation of Calcium Carbonate in Artificial Seawater at Sustained ./Supersaturation. Environ. Technol. 1992, 13 (1), 73−80.
• Bischoff, J. L. …Catalysis, Inhibition, and the Calcite-Aragonite …Problem; [Part] 2, The Vaterite-Aragonite Transformation. Am. J. Sci. 1968, 266 (2), 80−90.
• Carino, A.; Testino, A.; Andalibi, M. R.; Pilger, F.; Bowen, P.; Ludwig, C. .7Thermodynamic-Kinetic Precipitation Modeling. A Case .7Study: The Amorphous Calcium Carbonate (ACC) Precipitation .7Pathway Unravelled. Cryst. Growth Des. 2017, 17 (4), 2006−2015.
• Parkhurst, D. L.; Appelo, C. A. J. .6Description of Input and
Examples for PHREEQC Version 3a Computer Program for .6Speciation, Batch-Reaction, One-Dimensional Transport, and Inverse .6Geochemical Calculations. US Geol. Surv. Technol. Methods Book 2013, 6, 497.
• Global CCS Institute. Characteristics of Large North American CO2 Point Sources. 2
https://hub.globalccsinstitute.com/publications/ 2building-cost-curves-co2-storage-north-america/22-characteristics- 2large-north-american (accessed April 10, 2019).
• SCRIPPS. The Keeling Curve. http://scripps.ucsd.edu/ programs/keelingcurve (accessed April 10, 2019).
• Porter, R. T. J.; Fairweather, M.; Pourkashanian, M.; Woolley, R.
M. …The Range and Level of Impurities in CO2 Streams from Different …Carbon Capture Sources. Int. J. Greenhouse Gas Control 2015, 36, 161− 174.
• Dieter, C. A.; Maupin, M. A.; Caldwell, R. R.; Harris, M. A.; Ivahnenko, T. I.; Lovelace, J. K.; Barber, N. L.; Linsey, K. S. Estimated Use of Water in the United States in 2015; Circular 1441; U.S. Geological Survey: Reston, VA, 2018; p 76.
• Qi, S. L.; Harris, A. C. Geochemical Database for the Brackish Groundwater Assessment of the United States; U.S. Geological Survey, 2017.
• Jones, E.; Qadir, M.; van Vliet, M. T. H.; Smakhtin, V.; Kang, S. …The State of Desalination and Brine Production: A Global Outlook. Sci. Total Environ. 2019, 657, 1343−1356.
• U.S. Geological Survey. Energy Resources Program.
https:// e n e r g y . u s g s . g o v / E n v i r o n m e n t a l A s p e c t s / EnvironmentalAspectsofEnergyProductionandUse/
ProducedWaters. aspx#3822110-overview (accessed April 4, 2018).
• Blondes, M.; Gans, K.; Engle, M.; Kharaka, Y.; Reidy, M.; Saraswathula, V.; Thordsen, J.; Rowan, E.; Morrissey, E. U.S. Geological Survey National Produced Waters Geochemical Database v2.3; U.S. Geological Survey, 2019.
• 2012−2023 Report on Global Caustic Soda Market Competition,
Status and Forecast, Market Size by Players, Regions, Type, Application, 2017.
–https://issuu.com/anujar/docs/10730302-2012-2023-report- –on-global (accessed January 2021).
• Lee, D.-Y.; Elgowainy, A. A.; Dai, Q. Life Cycle Greenhouse Gas Emissions of By-Product Hydrogen from Chlor-Alkali Plants; ANL/ ESD-17/27; Argonne National Laboratory, 2017.
• Food and Agriculture Organization of the United Nations. AQUASTAT: Water Withdrawal by Sector, around 2010, 2016.
http:// www.fao.org/nr/water/aquastat/tables/WorldData-Withdrawal_eng. pdf (accessed January 2021).
• U.S. Environmental Protection Agency. The Sources and Solutions: Wastewater.
epa.gov/nutrientpollution/sources-and-solutions-wastewater (accessed May 12, 2019).
• U.S. Environmental Protection Agency. Clean Watersheds Needs Survey 2012 Report to Congress; EPA-830-R-15005; U.S. Environmental Protection Agency, 2016.
• Metropolitan Water Reclamation District of Greater Chicago. Stickney Water Reclamation Plant: Fact Sheet; Metropolitan Water Reclamation District of Greater Chicago, 2019.
• Aguilar, S.; Bustillos, S.; Xue, S.; Ji, C.-H.; Mak, W. H.; Rao, E.; McVerry, B. T.; La Plante, E. C.; Simonetti, D.; Sant, G.; Kaner, R. B. .0Enhancing Polyvalent Cation Rejection Using Perfluorophenylazide- .0Grafted-Copolymer Membrane Coatings. ACS Appl. Mater. Interfaces
2020, 12 (37), 42030−42040.
• Gabrielli, C.; Maurin, G.; Francy-Chausson, H.; Thery, P.; Tran,
T. T. M.; Tlili, M. …Electrochemical Water Softening: Principle and …Application. Desalination 2006, 201 (1), 150−163.
• Lei, Y.; Hidayat, I.; Saakes, M.; van der Weijden, R.; Buisman, C.
J. N. …Fate of Calcium, Magnesium and Inorganic Carbon in …Electrochemical Phosphorus Recovery from Domestic Wastewater. Chem. Eng. J. 2019, 362, 453−459.
• Keith, D. W.; Holmes, G.; St. Angelo, D.; Heidel, K. …A Process for …Capturing CO2 from the Atmosphere. Joule 2018, 2 (8), 1573−1594.
• Keith, D. W.; Ha-Duong, M.; Stolaroff, J. K. .–Climate Strategy .–with CO2 Capture from the Air. Clim. Change 2006, 74 (1), 17−45.
• Fujimura, K.; Izumiya, K.; Kawashima, A.; Akiyama, E.; Habazaki, H.; Kumagai, N.; Hashimoto, K. Anodically Deposited Manganese-Molybdenum Oxide Anodes with High Selectivity for Evolving Oxygen in Electrolysis of Seawater. J. Appl. Electrochem. 1999,
29 (6), 769−775.
• Bennett, J. E. ./-()Electrodes for Generation of Hydrogen and Oxygen ./-()from Seawater. Int. J. Hydrogen Energy 1980, 5 (4), 401−408.
• Lin, H.-W.; Cejudo-Marín, R.; Jeremiasse, A. W.; Rabaey, K.; Yuan, Z.; Pikaar, I. .Direct Anodic Hydrochloric Acid and Cathodic .Caustic Production during Water Electrolysis. Sci. Rep. 2016, 6, 1−4.
• Díaz, E.; Ordóñez, S.; Vega, A.; Coca, J. …Adsorption
Characterisation of Different Volatile Organic Compounds over …Alumina, Zeolites and Activated Carbon Using Inverse Gas …Chromatography. J. Chromatogr. A 2004, 1049 (1), 139−146.
• Liu, E.; Sarkar, B.; Chen, Z.; Naidu, R. …Decontamination of
Chlorine Gas by Organic Amine Modified Copper-Exchanged Zeolite.
Microporous Mesoporous Mater. 2016, 225, 450−455.
• De Yoreo, J. J.; Vekilov, P. G. ./Principles of Crystal Nucleation ./and Growth. Rev. Mineral. Geochem. 2003, 54 (1), 57−93.
• Dudchenko, A. V.; Chen, C.; Cardenas, A.; Rolf, J.; Jassby, D. ..Frequency-Dependent Stability of CNT Joule Heaters in Ionizable ..Media and Desalination Processes. Nat. Nanotechnol. 2017, 12 (6),
557−563.
• Gandía, L. M.; Arzamendi, G.; Dieǵuez, P. M. Renewable
Hydrogen Technologies; Elsevier, 2013. .DOI: 10.1002/ente.201402151.
• Hashaikeh, R.; Lalia, B. S.; Kochkodan, V.; Hilal, N. …A Novel in …Situ Membrane Cleaning Method Using Periodic Electrolysis. J. Membr. Sci. 2014, 471, 149−154.
• Hu, X.; Tian, X.; Lin, Y.-W.; Wang, Z. .9Nickel Foam and .9Stainless Steel Mesh as Electrocatalysts for Hydrogen Evolution .9Reaction, Oxygen Evolution Reaction and Overall Water Splitting in .9Alkaline Media. RSC Adv. 2019, 9 (54), 31563−31571.
• Elimelech, M.; Phillip, W. A. .The Future of Seawater .Desalination: Energy, Technology, and the Environment. Science 2011, 333 (6043), 712−717.
• Bartels, C. R.; Andes, K. ./..Consideration of Energy Savings in ./..SWRO. Desalin. Water Treat. 2013, 51 (4−6), 717−725.
• Pearce, G. K. …UF/MF Pre-Treatment to RO in Seawater and …Wastewater Reuse Applications: A Comparison of Energy Costs. Desalination 2008, 222 (1), 66−73.
• BHADLA, Climate Investment Funds. Solar Parks Transform India’s Energy Landscape. https://www.climateinvestmentfunds.org/ CIF10/india/bhadla (accessed April 7, 2020).
• U.S. Energy Information Administration. Annual Energy Outlook 2020; U.S. Energy Information Administration, 2020.
• Bureau of Economic Analysis. Gross Domestic Product, Fourth Quarter and Year 2019 (Advance Estimate); BEA 20-04; Bureau of Economic Analysis, 2018
• International Monetary Fund. Real GDP Growth.
NGDP_RPCH@WEO/OEMDC/
ADVEC/WEOWORLD
(accessed March 1, 2020).
• U.S. Department of Energy, Office of Energy Efficiency &
Renewable Energy. The Sun Shot Initiative. https://www.energy.gov/ eere/solar/sunshot-initiative (accessed Jul 23, 2019).
• California ISO. Price Map http://www.caiso.com/ TodaysOutlook/Pages/prices.aspx (accessed April 7, 2020).
• International Energy Agency, Photovoltaic Power Systems Technology Collaboration Programme. 2019 Snapshot of Global PV Markets; IEA PVPS T1-35:2019; International Energy Agency, 2019.
• Feldman, D.; Margolis, R. Q4 2018/Q1 2019 Solar Industry Update; NREL/PR-6A20-73992; National Renewable Energy Labo- ratory, 2019.
• Rau, G. H.; Willauer, H. D.; Ren, Z. J. .—The Global Potential for .—Converting Renewable Electricity to Negative-CO2 -Emissions Hydro- .—gen. Nat. Clim. Change 2018, 8 (7), 621−625.
• International Energy Agency. World Energy Outlook 2019. https://www.iea.org/reports/world-energy-outlook-2019 (accessed April 1, 2020).
• Köhler, P.; Hartmann, J.; Wolf-Gladrow, D. A. .Geoengineering
Potential of Artificially Enhanced Silicate Weathering of Olivine. Proc. Natl. Acad. Sci. U. S. A. 2010, 107 (47), 20228−20233.
• Cao, L.; Caldeira, K. ./-///Atmospheric Carbon Dioxide Removal: ./-///Long-Term Consequences and Commitment. Environ. Res. Lett. 2010, 5 (2), 024011.
• National Centers for Environmental Information, National Oceanic and Atmospheric Administration. State of the Climate: Global Climate Report for April 2020; National Centers for Environmental Information, 2020.
.ncdc.noaa.gov/sotc/global/202004
(accessed January 2021).
• United States Geological Survey. Limestone: A Crucial and Versatile Industrial Mineral Commodity. /https://pubs.usgs.gov/fs/ /2008/3089/ (accessed April 4, 2018).
• U.S. Geological Survey. Minerals Information: Crushed Stone.
minerals.usgs.gov/minerals/pubs/
commodity/stone_crushed/ (accessed April 3, 2018).
• Kaza, S.; Yao, L.; Bhada-Tata, P.; Van Woerden, F. What a Waste 2.0: A Global Snapshot of Solid Waste Management to 2050; Urban Development Series; World Bank: Washington, DC, 2018.
• World Bank. What a Waste Global Database. https:// datacatalog.worldbank.org/dataset/what-waste-global-database (ac- cessed April 25, 2019).
• U.S. Environmental Protection Agency. Municipal Solid Waste Landfills: Economic Impact Analysis for the Proposed New Subpart to the New Source Performance Standards, 2014. –https://beta.regulations.gov/ –document/EPA-HQ-OAR-2003-0215-0045 (accessed January 2021).
• United Nations Environment Programme; International Resource Panel. Global Material Flows and Resource Productivity:
Assessment Report for the UNEP International Resource Panel, 2016.
https://www.resourcepanel.org/reports/global-material-flows-and- resource-productivity-database-link (accessed January 2021).
• Materialflows.net. Data Visualisations. http://www. materialflows.net/visualisation-centre/data-visualisations/ accessed January 2021).
• Marufuzzaman, M.; Eksi̧oğlu, S. D.; Hernandez, R. …Truck versus
Pipeline Transportation Cost Analysis of Wastewater Sludge. Transp. Res. Part Policy Pract. 2015, 74, 14−30.
• U.S. Environmental Protection Agency. 1996 Protocol to the Convention on the Prevention of Marine Pollution by Dumping of Wastes and Other Matter, 1972 (as amended in 2006).
epa.gov/ocean-dumping/1996-protocol-convention-prevention-marine- pollution-dumping-wastes-and-other-matter
(accessed January 2021).
• Pytkowicz, R. M. ./-()-On the Carbonate Compensation Depth in ./-()-the Pacific Ocean. Geochim. Cosmochim. Acta 1970, 34 (7), 836−839.
• Feely, R. A.; Sabine, C. L.; Byrne, R. H.; Millero, F. J.; Dickson,
A. G.; Wanninkhof, R.; Murata, A.; Miller, L. A.; Greeley, D. ./Decadal ./Changes in the Aragonite and Calcite Saturation State of the Pacific ./Ocean. Global Biogeochem. Cycles 2012, 26 (3), 1−15.
• Caldeira, K.; Rau, G. H. ./Accelerating Carbonate Dissolution to ./Sequester Carbon Dioxide in the Ocean: Geochemical Implications. Geophys. Res. Lett. 2000, 27 (2), 225−228.
• Harvey, L. D. D. ./Mitigating the Atmospheric CO2 Increase and ./Ocean Acidification by Adding Limestone Powder to Upwelling ./Regions. J. Geophys. Res. 2008, 113 (C4), 1−21.
• Vitousek, S.; Barnard, P. L.; Limber, P.; Erikson, L.; Cole, B. ./A ./Model Integrating Longshore and Cross-Shore Processes for Predicting ./Long-Term Shoreline Response to Climate Change. J. Geophys. Res. Earth Surf. 2017, 122 (4), 782−806.
• NOAA Office for Coastal Management. Shoreline Mileage of the
United States.
https://coast.noaa.gov/data/docs/states/shorelines.pdf (accessed January 2021.
• Legal Information Institute. 26 U.S. Code § 45Q: Credit for
carbon oxide sequestration.
https://www.law.cornell.edu/uscode/text/ /26/45Q (accessed May 1, 2019).
• California Air Resources Board. Weekly LCFS Credit Trading Activity Reports.
https://www.arb.ca.gov/fuels/lcfs/credit/ lrtweeklycreditreports.htm (accessed May 1, 2019).
• Mehdipour, I.; Falzone, G.; La Plante, E. C.; Simonetti, D.; Neithalath, N.; Sant, G. .9How Microstructure and Pore Moisture Affect .9Strength Gain in Portlandite-Enriched Composites That Mineralize .9CO2. ACS Sustainable Chem. Eng. 2019, 7 (15), 13053−13061.
Proudly Supporting
Jim Lee at his site it is the most comprehensive
and up to date site on the net
https://climateviewer.org
